Key Takeaways
Autoimmune hemolytic anemia (AIHA) happens when the immune system mistakenly attacks and destroys the body's own red blood cells.
- Types ▾: There are two main forms; warm AIHA, driven by IgG antibodies active at body temperature, and cold agglutinin disease (CAD), driven by IgM antibodies active at cooler temperatures.
- Diagnosis ▾: Diagnosis relies on blood tests showing red blood cell breakdown plus a positive direct antiglobulin test (DAT).
- Treatment ▾: Warm AIHA is treated first with corticosteroids, then rituximab if needed; CAD does not respond to steroids and is instead managed with cold avoidance, rituximab, or complement inhibitors.
*Click ▾ for more information
What is Autoimmune Hemolytic Anemia?
Autoimmune hemolytic anemia (AIHA) is a rare blood disorder in which the immune system mistakes red blood cells for a threat and destroys them. It's rare, affecting an estimated 1 to 3 people per 100,000 each year, but it's an important condition to understand because it can develop quickly and become serious without treatment.
Red blood cells carry oxygen to every tissue in the body. In AIHA, the immune system produces antibodies that stick to these cells and mark them for destruction. This breakdown process is called hemolysis, and it happens faster than the bone marrow can replace the lost cells, leading to anemia (a shortage of healthy red blood cells).
Hemolysis can happen in two places:
- Intravascular hemolysis: red blood cells break apart directly inside blood vessels.
- Extravascular hemolysis: red blood cells are removed by the spleen and liver after being tagged by antibodies. This is the more common route in AIHA.
When red blood cells break down, three things typically follow: fewer working red blood cells (causing anemia symptoms like fatigue), a buildup of bilirubin from broken-down hemoglobin (causing jaundice, or yellowing of the skin and eyes), and, in severe cases, free hemoglobin spilling into the urine (hemoglobinuria).
Intravascular vs Extravascular Hemolysis
| Feature | Intravascular Hemolysis | Extravascular Hemolysis |
|---|---|---|
| Where it happens | Inside blood vessels | Mainly the spleen |
| Typical trigger | Severe infection, incompatible transfusion | Autoimmune disease, inherited red cell defects |
| Speed | Rapid, can cause shock | Usually more gradual |
| Example conditions | Severe transfusion reactions | Autoimmune hemolytic anemia, hereditary spherocytosis |
Types of AIHA
AIHA isn't one disease; it's a family of conditions grouped by which antibody is doing the damage and at what temperature it works best.
Warm AIHA is the most common type, making up about 70-80% of adult cases [1]. The antibodies (usually IgG) are active at normal body temperature (37°C). They coat red blood cells, and the spleen's macrophages, a type of immune cell that engulfs and digests targeted material, remove them.
Cold agglutinin disease (CAD) accounts for most of the remaining cases. Here, the antibodies (usually IgM) work best at cooler temperatures, generally below 30°C, which is why symptoms often show up in the fingers, toes, ears, and nose, where blood cools slightly as it circulates. These antibodies cause red blood cells to clump together (agglutinate), which can temporarily block small blood vessels.
Paroxysmal cold hemoglobinuria (PCH) is a rarer type where a specific "biphasic" antibody attaches to red blood cells in the cold and then triggers destruction once the blood warms back up. It's more common in children after a viral illness.
Mixed AIHA occurs when both warm and cold antibodies are active in the same person, producing features of both types.
Drug-induced immune hemolytic anemia (DIIHA) happens when a medication triggers antibody production against red blood cells; it usually resolves once the drug is stopped.
| Type | Antibody | Optimal Temperature | Main Site of Destruction | DAT Result |
|---|---|---|---|---|
| Warm AIHA | IgG | Body temperature | Spleen | Positive for IgG (± C3d) |
| Cold Agglutinin Disease | IgM | Below 30°C | Liver, plus inside vessels | Positive for C3d |
| Mixed AIHA | IgG and IgM | Both | Both | Positive for IgG and C3d |
| PCH | Biphasic IgG | Binds cold, destroys warm | Inside vessels | Positive for C3d only |
| Drug-Induced AIHA | IgG or IgM | Variable | Variable | Variable |
Causes of AIHA
About half of AIHA cases have no identifiable trigger. These are called primary or idiopathic AIHA.
The other half are secondary, meaning they develop alongside another condition, such as:
- Infections (Epstein-Barr virus, cytomegalovirus)
- Other autoimmune diseases (lupus, Sjögren's syndrome)
- Certain medications
- Cancers of the blood or lymph system (lymphoma, leukemia)
Pathophysiology of AIHA
Understanding the mechanism helps make sense of why treatments differ between warm and cold AIHA.

In warm AIHA: B lymphocytes produce IgG antibodies against red blood cell surface proteins. These antibodies stick to the red blood cells, and macrophages in the spleen, which have receptors that recognize the "tail" end of an antibody, grab and destroy the coated cells. Sometimes the spleen only removes part of the cell membrane rather than the whole cell; when that happens, the red blood cell shrinks into a smaller, sphere-shaped cell called a spherocyte, a classic clue seen under the microscope in warm AIHA.
IgG can also activate the complement system, a chain of about 20 blood proteins that normally helps clear infections. When complement is triggered, proteins called C3b and C4b stick to the red blood cell surface, adding an extra "destroy me" tag that also draws in macrophages.

In cold agglutinin disease: IgM antibodies bind red blood cells at cooler temperatures and activate complement too, but less efficiently than IgG does. This weaker activation usually stops short of directly bursting the cell and instead deposits C3b on the surface, marking it for removal by the liver and spleen. The IgM antibody itself often falls off once the blood warms back up, leaving only the complement marker behind, which is why cold agglutinin disease often shows a different DAT pattern than warm AIHA.
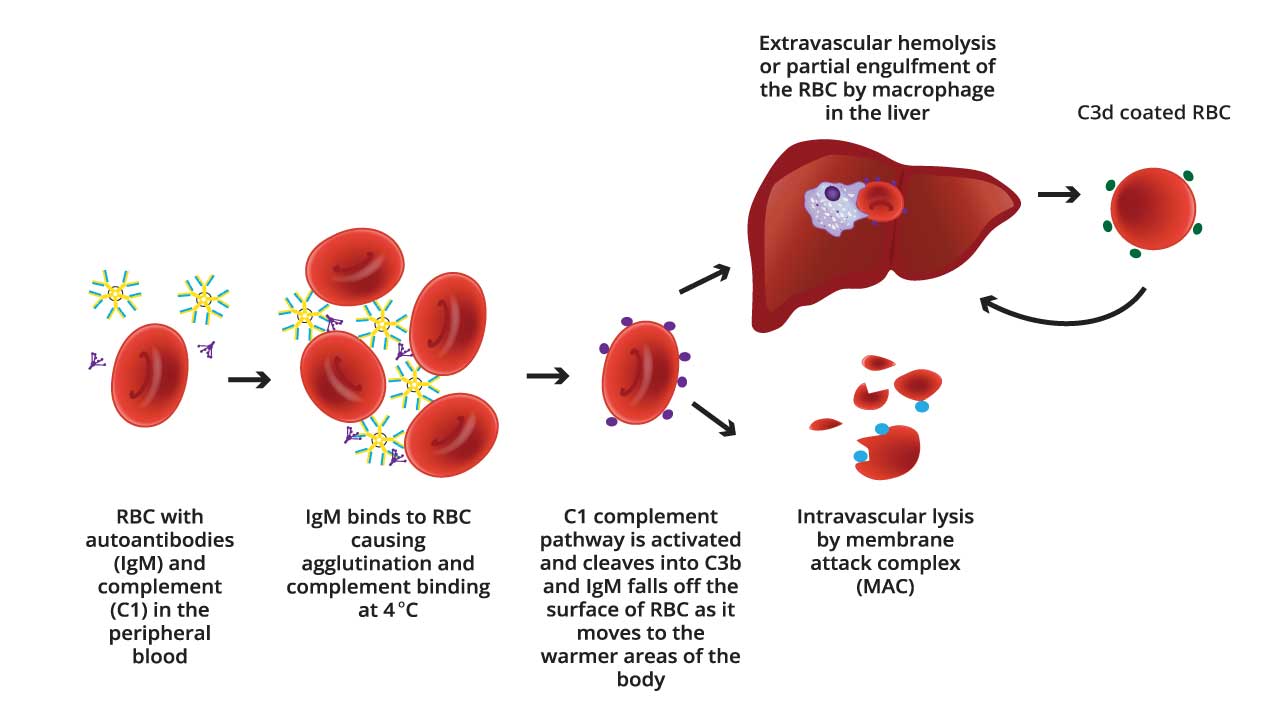
The complement system is normally kept in check by several regulatory proteins on healthy cells (such as CD59 and Factor H) that stop it from attacking the body's own tissue. In autoimmune disease, this balance breaks down, allowing complement to be activated against red blood cells that should otherwise be protected.
Signs and Symptoms
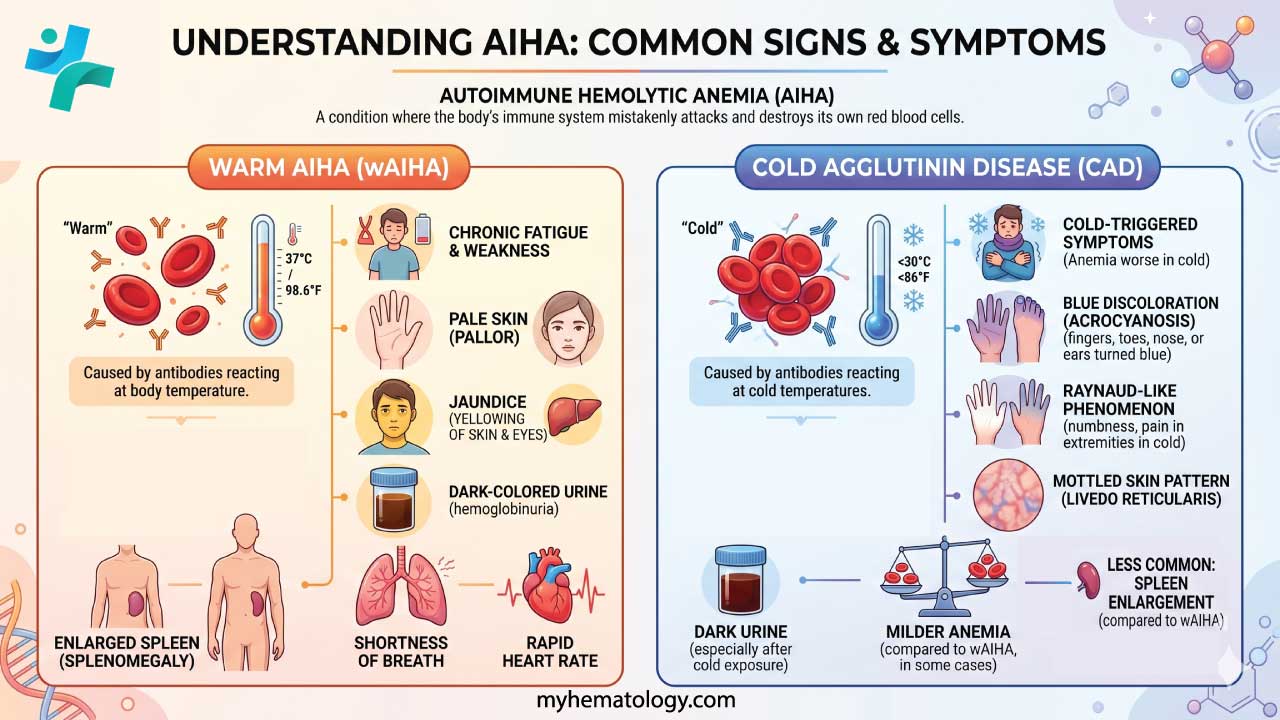
Because anemia is the core problem, most symptoms come from the body not getting enough oxygen:
- Fatigue and weakness
- Shortness of breath
- Pale skin
- Dizziness or lightheadedness
- Rapid heartbeat
Additional symptoms come from the breakdown products of destroyed red blood cells:
- Jaundice: yellowing of the skin and the whites of the eyes, from bilirubin buildup
- Dark urine: from hemoglobin breakdown products
- Splenomegaly: an enlarged spleen, sometimes felt on physical exam
CAD has some symptoms of its own, caused by clumped red blood cells slowing blood flow in cooler areas:
- Acrocyanosis: bluish discoloration of the fingers, toes, or ears in the cold
- Livedo reticularis: a lacy, purple-red pattern on the skin, usually on the legs
Complications
Left untreated, AIHA can lead to:
- Severe anemia, causing extreme fatigue and, in serious cases, heart strain from the heart working harder to deliver oxygen
- Increased infection risk, partly from the disease itself and partly from immune-suppressing treatments
- Blood clots (thrombosis), including deep vein thrombosis and pulmonary embolism; this risk is more pronounced in warm AIHA
- Skin ulceration or gangrene in severe CAD, from prolonged blood flow blockage in the extremities
Diagnosis and Laboratory Investigations
Diagnosing AIHA follows three steps: confirm hemolysis is happening, confirm it's immune-related, and determine whether it's warm or cold.

Step 1: Confirm hemolysis.
- Reticulocyte count: usually high, as the bone marrow works overtime to replace lost cells
- Haptoglobin: a protein that mops up free hemoglobin; it drops low or becomes undetectable during active hemolysis
- Lactate dehydrogenase (LDH): an enzyme released from broken-down cells; levels rise
- Unconjugated bilirubin: rises as hemoglobin is broken down
- Urinalysis: may show hemoglobin or hemosiderin in more severe, intravascular cases
Step 2: Look at the blood smear. Warm AIHA typically shows spherocytes (small, round red blood cells missing their normal central pale area). CAD shows red blood cells clumped together in irregular clusters.
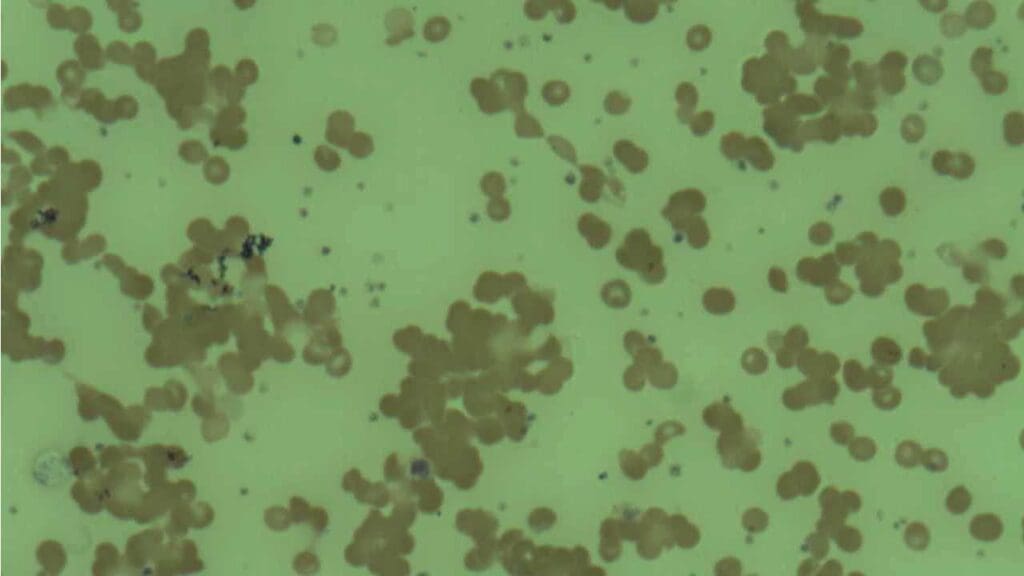
Step 3: Confirm it's immune-mediated with the direct antiglobulin test (DAT). This is the single most important test. A positive DAT for IgG points to warm AIHA; a positive DAT for complement (C3d) alone points more toward CAD or PCH. In about 3-5% of clinically clear AIHA cases, the standard DAT comes back negative. This typically occurs because the autoantibodies have a low binding affinity, are present in quantities too small for standard reagents to detect, or belong to the IgA class rather than IgG or IgM. A negative DAT does not rule out AIHA if active hemolysis is clinically evident; in these cases, more sensitive tests such as microcolumn gel tests, flow cytometry, or specific testing for IgA autoantibodies are required [1].
Additional tests may include a cold agglutinin titer (a level of 64 or higher at 4°C supports CAD) and, for suspected PCH, the Donath-Landsteiner test, which looks for the biphasic antibody characteristic of that condition.
Additionally, for patients suspected of having Cold Agglutinin Disease, current diagnostic guidelines strongly recommend a bone marrow biopsy. Because CAD is frequently driven by an underlying low-grade clonal B-cell lymphoproliferative disorder (such as a marginal zone lymphoma), evaluating the bone marrow is a critical step that helps direct long-term treatment and management [11].
Ruling Out Other Conditions
Several other disorders can look like AIHA at first glance, so doctors typically rule these out during workup:
| Condition | Key Clue | DAT Result |
|---|---|---|
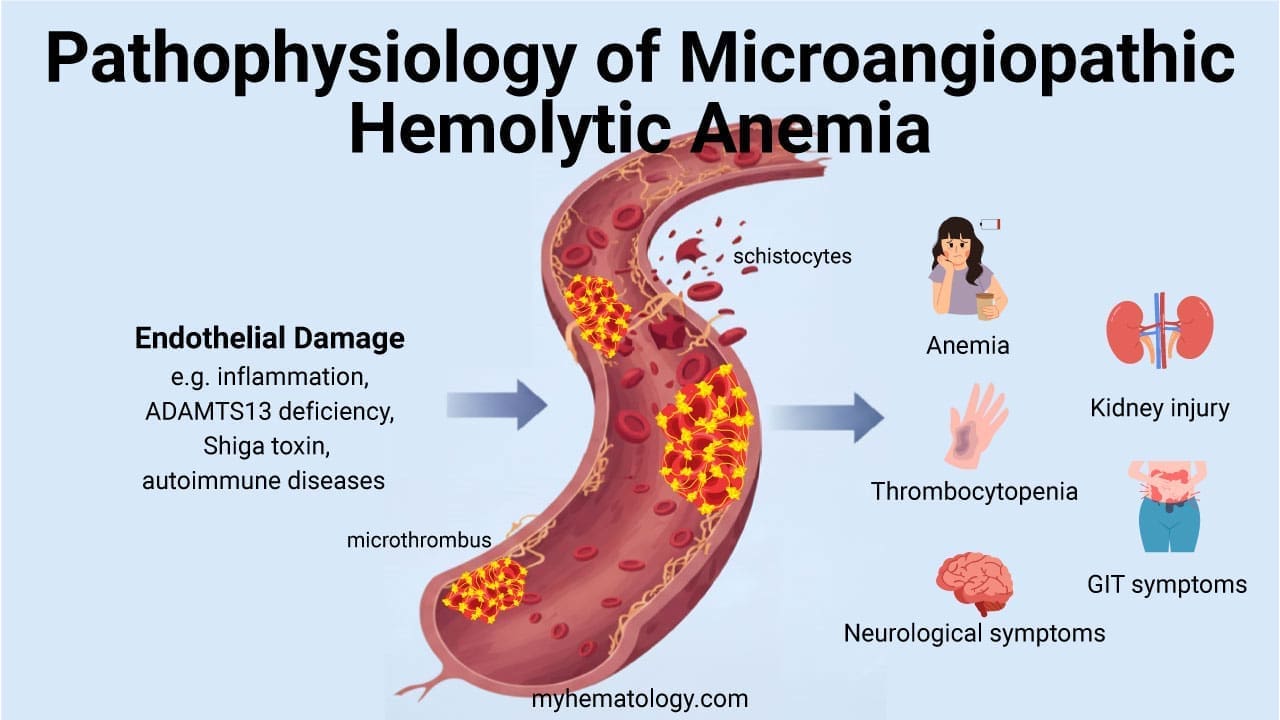
| Microangiopathic hemolytic anemia (e.g., TTP, HUS) | Fragmented red cells (schistocytes) on smear, low platelets | Negative |
| Hereditary spherocytosis | Family history, spherocytes present | Negative |
| G6PD deficiency | Recent oxidant drug or fava bean exposure | Negative |
| Paroxysmal nocturnal hemoglobinuria | Morning dark urine, unusual clot locations | Usually negative |
| Alloimmune hemolysis | Recent transfusion or pregnancy | Positive |
Treatment and Management of AIHA
Treatment depends heavily on whether the antibody is warm (IgG) or cold (IgM), because the two forms respond very differently to the same drugs.
- First-line: corticosteroids. Prednisone (typically 1-1.5 mg/kg per day) reduces macrophage activity against coated red blood cells and lowers antibody production over time. Once hemoglobin rises above 10 g/dL, the dose is tapered slowly over 3-6 months to reduce relapse risk [7].
- Second-line: rituximab. This antibody targets the B cells making the harmful autoantibodies and is now often used earlier for patients who don't respond well to steroids or need high maintenance doses [1].
- Third-line and refractory options: splenectomy (surgical removal of the spleen), or steroid-sparing immunosuppressants such as azathioprine, mycophenolate mofetil, or cyclosporine.
CAD behaves differently: corticosteroids and splenectomy generally don't work, because the disease process depends more on complement activation than on spleen-based antibody clearance.
- Thermal protection is the most important everyday intervention: keeping hands, feet, and ears warm at all times.
- Blood warmers are used during any transfusion to stop cold antibodies from activating in the donor blood.
- Rituximab, often combined with bendamustine, is considered first-line drug therapy [1].
- Sutimlimab (brand name Enjaymo), FDA-approved in 2022, blocks a specific complement protein (C1s) and switches off hemolysis without broadly suppressing the immune system. While sutimlimab rapidly halts hemolysis and is highly recommended for patients who cannot tolerate chemotherapy or require immediate stabilization, it requires continuous, ongoing infusions. For patients seeking deep, long-term disease remission, classical treatments (such as rituximab combined with bendamustine) often remain the favored first-line approach [11,12].
| Feature | Warm AIHA (IgG) | Cold Agglutinin Disease (IgM) |
|---|---|---|
| Main site of destruction | Spleen | Liver, plus inside vessels |
| Corticosteroids |
Highly effective
First-line
|
Not effective |
| Splenectomy |
Effective
Later-line
|
Not effective |
| Rituximab |
Effective
Second-line
|
Highly effective
First-line
|
| Complement inhibitors | Rarely used |
FDA-approved
Sutimlimab
|
| Cold avoidance | Not required | Essential |
Recent developments to watch: The treatment landscape for warm AIHA is shifting rapidly toward targeted therapies. In early 2026, the FDA granted Priority Review for nipocalimab (an immunoselective drug that blocks the neonatal Fc receptor (FcRn) to rapidly clear pathogenic IgG autoantibodies) positioning it to potentially become the first-ever approved treatment dedicated to warm AIHA [10]. Simultaneously, rilzabrutinib, an oral Bruton’s tyrosine kinase (BTK) inhibitor, received FDA Breakthrough Therapy designation. Rilzabrutinib represents a highly promising emerging option for patients who do not respond to standard steroids or rituximab [9].
Emergency and supportive care, regardless of type:
- Transfusion: because the autoantibody often reacts against most donor blood, crossmatching frequently shows "incompatibility." The clinical rule is to never withhold blood in life-threatening anemia; doctors use the "least incompatible" units available and monitor closely.
- Blood clot prevention: active hemolysis is a strongly clot-promoting state. Hospitalized patients with active AIHA are often given preventive blood thinners (such as low molecular weight heparin) unless there's a reason not to.
- Folate supplementation: chronic hemolysis increases the demand for folate as the bone marrow works harder to produce new red blood cells; 1-5 mg of folic acid daily helps prevent a folate-deficiency crisis.
Memory aids
Warm AIHA – the 3 S's: Steroids (first-line) · Splenectomy (for refractory cases) · Spherocytes (seen on the smear)
CAD – the 3 C's: Cold avoidance · Complement (C3d) · C1s inhibitors like sutimlimab
Prognosis
Outcomes vary by type, severity, cause, and how well someone responds to treatment.
Warm AIHA has a more variable course. Favorable signs include early diagnosis, good response to steroids or rituximab, and successful treatment of any underlying cause. Unfavorable signs include delayed diagnosis, severe anemia needing repeated transfusions, and relapse despite treatment.
CAD generally has a more favorable course than warm AIHA, especially with consistent cold avoidance and treatment response.
One large Danish cohort study found a median survival of 9.8 years for primary AIHA and 3.3 years for secondary AIHA after diagnosis [6].
Long-term management for both types typically involves regular monitoring and, for some patients, ongoing low-dose immunosuppression to prevent relapse.
Frequently Asked Questions (FAQs)
What is autoimmune hemolytic anemia?
Autoimmune hemolytic anemia (AIHA) is a rare condition where the immune system mistakenly makes antibodies against a person's own red blood cells, causing them to break down faster than the body can replace them. This leads to anemia and its associated symptoms, like fatigue and shortness of breath.
What's the difference between warm and cold AIHA?
Warm AIHA involves IgG antibodies that attack red blood cells at normal body temperature, with the spleen doing most of the destroying. Cold agglutinin disease involves IgM antibodies that are more active at cooler temperatures, causing red blood cells to clump in cooler areas like the fingers and toes.
How is AIHA diagnosed?
Diagnosis relies on blood tests showing active red blood cell breakdown (low haptoglobin, high LDH, high bilirubin, high reticulocyte count), combined with a positive direct antiglobulin test (DAT), which confirms the destruction is immune-related.
Is AIHA curable?
It depends on the cause. Secondary AIHA, triggered by an infection, medication, or another disease, can sometimes resolve completely once the trigger is treated. Primary AIHA tends to be chronic and is managed long-term, though many people reach periods of remission.
Can someone with AIHA safely receive a blood transfusion?
Yes, though it takes extra care. Because antibodies coat the red blood cells generally, crossmatching donor blood often looks "incompatible" in the lab. In severe, life-threatening anemia, doctors use the "least incompatible" blood available, since correcting dangerous oxygen shortage outweighs the transfusion risk.
Why are people with AIHA at higher risk of blood clots?
Active hemolysis releases substances that promote clotting and reduces a molecule (nitric oxide) that normally helps blood vessels relax. This is why hospitalized patients with active AIHA are often given preventive blood thinners.
Glossary of Related Medical Terms
- Agglutination: Clumping together of red blood cells, seen in cold agglutinin disease.
- Antibody: A protein made by the immune system to target foreign invaders; in AIHA it mistakenly targets the body's own red blood cells.
- Complement system: A group of blood proteins that work together to help destroy targeted cells.
- Direct antiglobulin test (DAT): A blood test that detects antibodies or complement stuck to red blood cells; the key confirmatory test for AIHA.
- Extravascular hemolysis: Red blood cell destruction outside the bloodstream, mainly by the spleen.
- Hemolysis: The breakdown of red blood cells before their normal lifespan is over.
- Intravascular hemolysis: Red blood cell destruction that happens directly inside blood vessels.
- Reticulocyte: A young, immature red blood cell; a high count shows the bone marrow working overtime.
- Spherocyte: A sphere-shaped red blood cell formed after the spleen removes part of its membrane; a classic sign of warm AIHA.
- Splenomegaly: Enlargement of the spleen.
- Thrombocytopenia: A lower-than-normal platelet count.
Disclaimer: This article is intended for informational purposes only and is specifically targeted towards medical students. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Jäger, U., Barcellini, W., Broome, C. M., Gertz, M. A., Hill, A., Hill, Q. A., Jilma, B., Kuter, D. J., Michel, M., Montillo, M., Röth, A., Zeerleder, S. S., & Berentsen, S. (2020). Diagnosis and treatment of autoimmune hemolytic anemia in adults: Recommendations from the First International Consensus Meeting. Blood reviews, 41, 100648. https://doi.org/10.1016/j.blre.2019.100648
- Deng, J., Zhou, F., Wong, C. Y., Huang, E., & Zheng, E. (2020). Efficacy of therapeutic plasma exchange for treatment of autoimmune hemolytic anemia: A systematic review and meta-analysis of randomized controlled trials. Journal of clinical apheresis, 35(4), 294–306. https://doi.org/10.1002/jca.21790
- Swiecicki, P. L., Hegerova, L. T., & Gertz, M. A. (2013). Cold agglutinin disease. Blood, 122(7), 1114–1121. https://doi.org/10.1182/blood-2013-02-474437
- Go, R. S., Winters, J. L., & Kay, N. E. (2017). How I treat autoimmune hemolytic anemia. Blood, 129(22), 2971–2979. https://doi.org/10.1182/blood-2016-11-693689
- Hill, A., & Hill, Q. A. (2018). Autoimmune hemolytic anemia. Hematology. American Society of Hematology. Education Program, 2018(1), 382–389. https://doi.org/10.1182/asheducation-2018.1.382
- Hansen, D. L., Möller, S., & Frederiksen, H. (2022). Survival in autoimmune hemolytic anemia remains poor, results from a nationwide cohort with 37 years of follow-up. European journal of haematology, 109(1), 10–20. https://doi.org/10.1111/ejh.13764
- Barcellini, W., & Fattizzo, B. (2021). How I treat warm autoimmune hemolytic anemia. Blood, 137(10), 1283–1294. https://doi.org/10.1182/blood.2019003808
- Berentsen S. (2025). Treatment of autoimmune hemolytic anemia: novel and investigational approaches. Minerva medica, 116(3), 249–264. https://doi.org/10.23736/S0026-4806.25.09617-X
- Sanofi. (2026, February 9). Sanofi's rilzabrutinib designated breakthrough therapy in the US and orphan drug in Japan for the treatment of warm autoimmune hemolytic anemia [Press release].
- Johnson & Johnson. (2026, February 24). Johnson & Johnson seeks FDA approval of IMAAVY (nipocalimab-aahu) as the first-ever FDA-approved treatment for warm autoimmune hemolytic anemia [Press release].
- Berentsen S. (2021). How I treat cold agglutinin disease. Blood, 137(10), 1295–1303. https://doi.org/10.1182/blood.2019003809
- Röth, A., Barcellini, W., D'Sa, S., Miyakawa, Y., Broome, C. M., Michel, M., Kuter, D. J., Jilma, B., Tvedt, T. H. A., Fröbel, J., Mastey, V., Nazir, S., Jiang, X., Shaheen, M., Hobbs, W., & Berentsen, S. (2022). Sutimlimab in patients with cold agglutinin disease: Results of the randomized placebo-controlled phase 3 CADENZA trial. Blood, 140(9), 980–991. https://doi.org/10.1182/blood.2021014955