Key Takeaways
Hemolytic anemia is a blood disorder where red blood cells (RBCs) are destroyed faster than they can be replaced.
- Types ▾:
- Hereditary: Caused by genetic mutations affecting RBC structure, function, or enzyme production (e.g., sickle cell disease, thalassemia).
- Acquired: Develops later in life due to various factors (e.g., autoimmune hemolytic anemia, drug-induced immune hemolytic anemia, infections).
- Symptoms ▾:
- Fatigue and weakness
- Pallor (pale skin)
- Jaundice (yellowing of skin and eyes)
- Dark urine (in some cases)
- Shortness of breath
- Rapid heart rate
- Enlarged spleen (in some cases)
- Complications ▾:
- Gallstones
- Increased risk of infections
- Leg ulcers (sickle cell disease)
- Heart problems
- Delayed growth and development (in children)
- Diagnosis ▾:
- Medical history and physical examination
- Blood tests (CBC, peripheral blood smear, DAT, haptoglobin, bilirubin)
- Additional tests specific to suspected cause (e.g., hemoglobin electrophoresis for sickle cell disease)
- Treatment ▾:
- Address the underlying cause (if possible)
- Supportive care (folic acid)
- Blood transfusions (if needed)
- Medications (e.g., immunosuppressants for autoimmune hemolysis)
- Procedures (e.g., splenectomy in some cases)
- Lifestyle modifications (depending on cause)
*Click ▾ for more information
Introduction
Hemolytic anemia is a blood disorder where red blood cells (RBCs) are destroyed faster than the body can replace them. This rapid or premature destruction reduces the availability of enough red blood cells for the normal physiological function of the body, leading to anemia with various symptoms and complications.
Normal RBC function and lifespan
Red blood cells, also known as erythrocytes, play a critical role in the body's oxygen transport system.
Function
- Oxygen Delivery: Red blood cells contain a protein called hemoglobin, which acts as a carrier for oxygen. Hemoglobin binds to oxygen in the lungs and transports it throughout the body.
- Carbon Dioxide Removal: They also play a role in removing carbon dioxide, a waste product of cellular respiration. Hemoglobin picks up carbon dioxide from tissues and carries it back to the lungs for exhalation.
Lifespan
- Production: Red blood cells are constantly produced in the bone marrow. This process, called erythropoiesis, takes about 7 days.
- Circulation: Mature red blood cells are released into the bloodstream and circulate for an average lifespan of 100-120 days. They lack a nucleus, allowing them to be flexible and squeeze through tiny blood vessels to deliver oxygen effectively.
- Destruction: As red blood cells age and become less efficient, they are removed from circulation by specialized cells called macrophages in the spleen and liver. The components of the destroyed red blood cells are then recycled by the body to create new ones.
What is extravascular hemolysis?
Extravascular hemolysis refers to the destruction of red blood cells (RBCs) outside the bloodstream, primarily occurring in the spleen and liver.

Location
- Spleen: This is the main site for extravascular hemolysis. The spleen has macrophages (white blood cells) that identify and remove old, damaged, or abnormal red blood cells from circulation.
- Liver: To a lesser extent, the liver can also remove damaged RBCs.
Process
- Targeting: Macrophages in the spleen and liver have receptors that recognize specific markers on the surface of red blood cells. These markers can be:
- Age-related changes: As RBCs age, their membranes become slightly altered, and macrophages recognize these changes.
- Abnormal shape or structure: Inherited disorders like sickle cell disease can cause RBCs to be abnormally shaped, making them more susceptible to destruction by macrophages.
- Antibody coating: In some cases, red blood cells may be coated with antibodies (proteins from the immune system). Macrophages have receptors that recognize these antibodies and target the coated RBCs for destruction.
- Phagocytosis: Once a macrophage identifies a target RBC, it engulfs and digests the cell, breaking down its components.
- Components: The breakdown products of the destroyed RBC, including hemoglobin, are released by the macrophages and processed further by the body. Hemoglobin, for example, is broken down into bilirubin, a yellow pigment that contributes to jaundice (yellowing of the skin) when present in high levels.
Examples of conditions that can cause extravascular hemolysis
- Hereditary spherocytosis
- Enzyme deficiencies
- Autoimmune hemolytic anemia (warm type)
What is intravascular hemolysis?
Intravascular hemolysis describes the destruction of red blood cells (RBCs) within the bloodstream itself. This rupturing of RBCs releases their contents, including hemoglobin, directly into the plasma, leading to different consequences compared to extravascular hemolysis.

Location
Intravascular hemolysis happens directly within blood vessels. This can occur throughout the circulatory system, but some areas are more prone to it due to mechanical stress on the RBCs, such as:
- Heart valves (especially damaged or prosthetic valves)
- Blood vessels with narrowed passages due to diseases like atherosclerosis
Process
- Direct Damage: RBCs are susceptible to damage from various factors within the bloodstream.
- Mechanical Stress: Shearing forces in turbulent blood flow can tear fragile RBCs, especially in conditions like prosthetic heart valves or narrowed blood vessels.
- Chemical or Toxin Exposure: Certain chemicals, toxins, or medications can directly damage the RBC membrane, leading to rupture. Examples include snake venom, certain antibiotics, and severe infections.
- Immune Attack: In some autoimmune hemolytic anemia cases (particularly cold autoimmune hemolytic anemia), antibodies target RBCs in the bloodstream, marking them for destruction by the immune system.
- Parasites: Certain parasites, like Babesia, can invade and lyse (rupture) RBCs from within.
- Hemolysis: When damage occurs, the RBC membrane ruptures, releasing its contents, including hemoglobin, into the bloodstream. Free-floating hemoglobin can cause further complications.
Consequences
- Hemoglobinemia: The presence of free hemoglobin in the plasma is called hemoglobinemia. This can overwhelm the body's natural processes for handling hemoglobin breakdown products.
- Hemosiderinuria: Free hemoglobin can be filtered by the kidneys, but it can damage them in the process. When excessive amounts are filtered, it can lead to hemosiderinuria, the presence of hemosiderin (iron from broken down hemoglobin) in the urine.
- Increased Bilirubin: Hemoglobin breakdown leads to the formation of bilirubin. Normally, the liver processes bilirubin, but in severe intravascular hemolysis, the liver may become overwhelmed, leading to high bilirubin levels and jaundice (yellowing of the skin and eyes).
Examples of conditions that can cause intravascular hemolysis
- Severe infections (sepsis)
- Autoimmune hemolytic anemia (cold type)
- Transfusion reaction (incompatibility between donor and recipient blood)
- Hemolytic uremic syndrome (HUS)
- Certain medications
- Prosthetic heart valves
Comparison of Extravascular vs. Intravascular Hemolysis
| Feature | Extravascular Hemolysis | Intravascular Hemolysis |
| Site of Destruction | Outside the blood vessels (primarily in the Spleen and Liver). | Directly within the circulating blood. |
| Primary Mechanism | Macrophages (splenic/hepatic) phagocytose "marked" or rigid RBCs. | Direct membrane rupture (complement, toxins, or mechanical shearing). |
| Hemoglobin Processing | Hemoglobin is broken down inside macrophages into heme and globin. | Free hemoglobin is released directly into the plasma. |
| Hemoglobinemia | Absent or very minimal. | Present (high levels of free Hb in the blood). |
| Hemoglobinuria | Absent (Hb is recycled locally). | Present (causes dark, "cola-colored" urine). |
| Hemosiderinuria | Absent. | Present (indicated by iron-stained sediment in urine; seen in chronic cases). |
| Haptoglobin Level | Decreased (but often less severely than intravascular). | Severely Decreased to Absent (depleted as it binds free Hb). |
| Serum Bilirubin | Significantly Increased (Unconjugated). | Increased (but often less than extravascular). |
| LDH Level | Increased. | Significantly Increased. |
| Abnormal RBC Morphology (Smear) | Often shows Spherocytes (remnants of partially eaten cells). | Often shows Schistocytes (fragments from mechanical damage). |
| Common Causes | Hereditary Spherocytosis, Warm AIHA, Thalassemia, Sickle Cell Disease. | G6PD Deficiency (acute), Cold Agglutinin Disease, PNH, MAHA (TTP/HUS), Mechanical Valves. |
| Splenomegaly | Common (due to increased work/hypertrophy). | Uncommon (destruction is systemic). |
Classification of Hemolytic Anemia Based on Cause
Hereditary hemolytic anemia
Hereditary hemolytic anemia is a group of blood disorders passed down through families (inherited) where there's a defect in the red blood cells (RBCs) themselves. This defect makes them more fragile and prone to premature destruction compared to healthy RBCs.
Pathophysiology
- Genetic Basis: Mutations in genes responsible for red blood cell structure, function, or enzyme activity are inherited from one or both parents.
- Defect in RBCs: These mutations lead to abnormalities in the RBC membrane, enzymes involved in red blood cell function, or the structure of hemoglobin (the oxygen-carrying protein within RBCs).
- Increased RBC Destruction: Due to the defect, RBCs become more susceptible to premature destruction by the spleen (extravascular hemolysis) or within the bloodstream (intravascular hemolysis).
- Symptoms: This increased destruction can lead to symptoms like fatigue, weakness, pallor (pale skin), jaundice (yellowing of the skin and eyes), and sometimes dark urine.
Types of Hereditary Hemolytic Anemia
Hereditary hemolytic anemias are classified based on the specific defect in the RBCs.
- Membrane Defects
- Hereditary spherocytosis: RBCs become sphere-shaped (spherocytes) and are more susceptible to destruction.
- Elliptocytosis: RBCs have an oval shape (elliptical) and may be less severe than spherocytosis.
- Enzyme Deficiencies:
- Glucose-6-phosphate dehydrogenase (G6PD) deficiency: This enzyme helps protect RBCs from oxidative damage. Deficiency makes them vulnerable to destruction, especially with certain triggers like medications or infections.
- Pyruvate kinase (PK) deficiency: This enzyme plays a role in RBC energy production. Deficiency can lead to decreased RBC lifespan.
- Hemoglobin Abnormalities:
- Sickle cell disease: A mutation in the hemoglobin gene causes RBCs to sickle (become crescent-shaped) and block blood vessels.
- Thalassemia: Mutations reduce or eliminate the production of globin chains, protein components of hemoglobin. This leads to abnormal hemoglobin and decreased RBC production.
Acquired hemolytic anemia
Acquired hemolytic anemia, unlike hereditary hemolytic anemia, is not something you're born with. It develops later in life due to various factors that damage or destroy healthy red blood cells (RBCs). This damage occurs outside of the inherent properties of the RBCs themselves.
Pathophysiology
The specific mechanism of RBC destruction depends on the cause. Here are some common scenarios:
- Antibody-mediated: In immune-mediated hemolytic anemia, antibodies attach to RBCs, marking them for destruction by macrophages in the spleen or liver.
- Complement activation: In some cases, the immune system may activate a protein cascade called the complement system, which directly damages the RBC membrane.
- Mechanical damage: In non-immune causes like red cell fragmentation syndromes, RBCs are physically damaged by shearing forces in the circulation.
- Chemical damage: Toxins or infections can directly damage the RBC membrane or alter their internal environment, leading to destruction.
Types of Acquired Hemolytic Anemia
A wide range of factors can trigger acquired hemolytic anemia. These can be broadly categorized as:
- Immune-mediated: The immune system mistakenly attacks healthy RBCs. This can be further classified as:
- Warm autoimmune hemolytic anemia (WAIHA): Antibodies target RBCs at body temperature.
- Cold agglutinin disease: Antibodies target RBCs at cooler temperatures.
- Alloimmune-related: Antibodies from a different blood type cause hemolysis, such as in a hemolytic transfusion reaction.
- Drug-induced: Certain medications can damage RBCs or trigger an immune reaction against them.
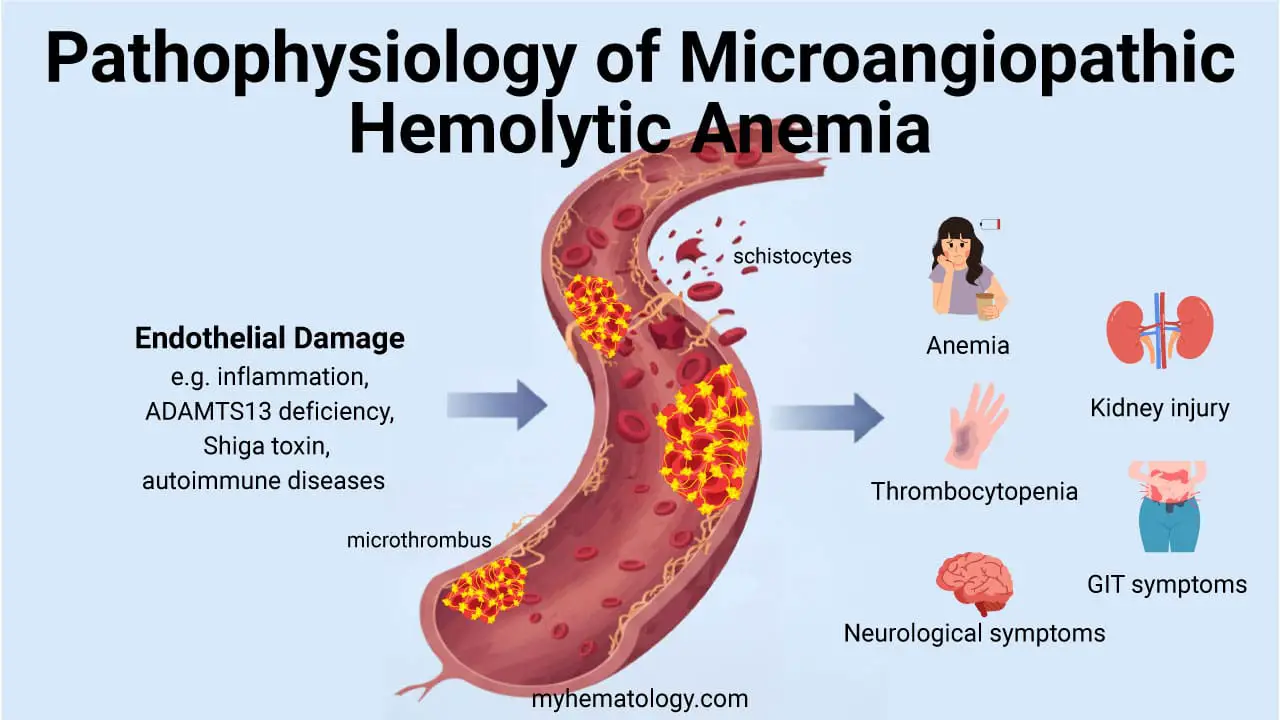
- Red cell fragmentation syndromes: Mechanical shearing forces fragment RBCs in the bloodstream (e.g., microangiopathic hemolytic anemia).
- Paroxysmal nocturnal hemoglobinuria (PNH): A rare blood disorder where RBCs are more susceptible to complement-mediated destruction.
- Infections: Some infections can damage RBCs or trigger an immune response leading to hemolysis.
- Chemical and physical agents: Severe burns, exposure to certain toxins, or snakebites can damage RBCs.
- Secondary causes: Underlying conditions like liver disease or severe kidney disease can contribute to hemolysis.
- March hemoglobinuria: A rare condition triggered by strenuous exercise in cold weather, causing RBC breakdown.
Classification of Hemolytic Anemia Based on Etiology
| Category | Sub-Category | Examples |
| Intrinsic (Hereditary) | Membrane Defects | Hereditary Spherocytosis, Hereditary Elliptocytosis. |
| Enzyme Deficiencies | G6PD Deficiency, Pyruvate Kinase (PK) Deficiency. | |
| Hemoglobinopathies | Sickle Cell Disease, Thalassemia. | |
| Membrane Protein Defects | Paroxysmal Nocturnal Hemoglobinuria (PNH)* | |
| Extrinsic (Acquired) | Immune-Mediated | Warm/Cold Autoimmune Hemolytic Anemia (AIHA), Alloimmune (Transfusion reactions), Drug-induced. |
| Mechanical (MAHA) | TTP, HUS, DIC, Prosthetic Heart Valves, March Hemoglobinuria. | |
| Infections | Malaria, Babesiosis, Clostridium perfringens (sepsis). | |
| Chemicals & Toxins | Snake/Spider venoms, Lead poisoning, Copper toxicity (Wilson’s Disease). | |
| Hypersplenism | Splenic sequestration (RBCs trapped in an enlarged spleen). |
* Note on PNH: While Paroxysmal Nocturnal Hemoglobinuria is an intrinsic defect (a mutation in the PIGA gene), it is an acquired clonal disorder rather than an inherited one.
Signs and Symptoms
Hemolytic anemia, whether inherited or acquired, disrupts the normal function of red blood cells (RBCs) by causing their premature destruction. This disruption leads to a range of signs and symptoms that can vary depending on the severity of the anemia.
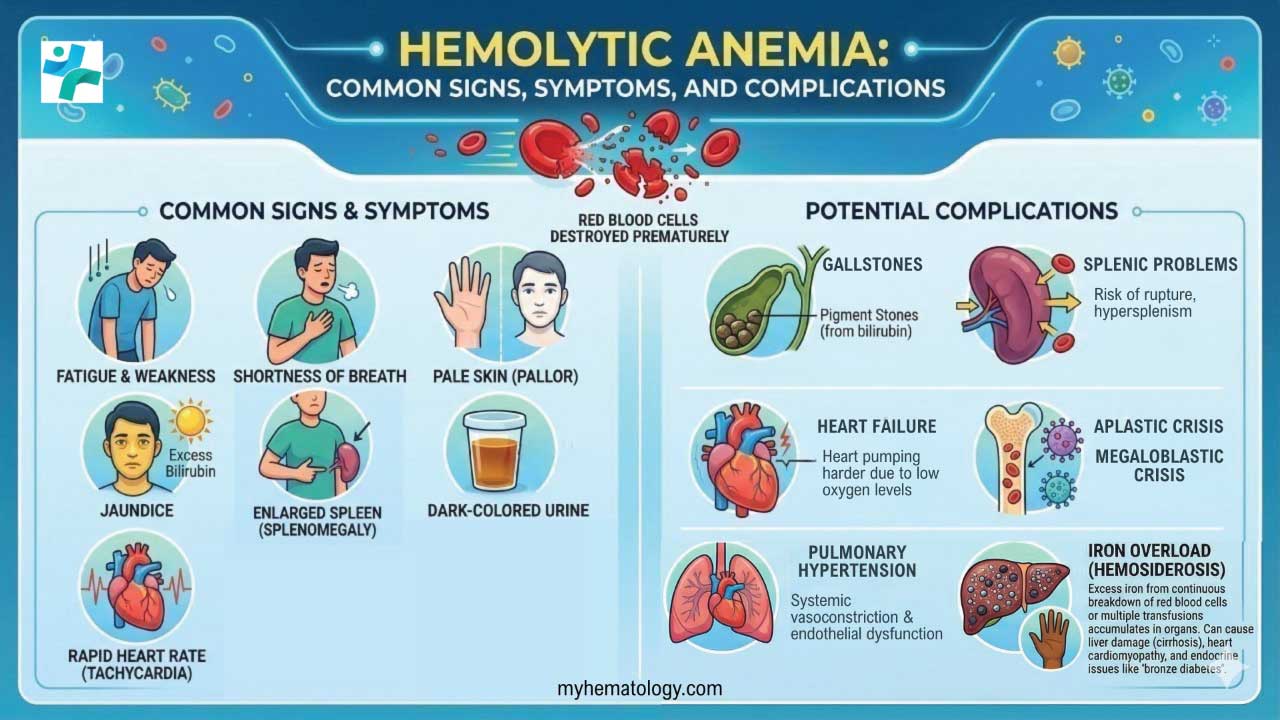
Common Symptoms
- Fatigue and weakness: This is a hallmark symptom due to reduced oxygen delivery to tissues throughout the body. People with hemolytic anemia may experience tiredness, difficulty concentrating, and a decreased ability to perform physical activities.
- Pallor (pale skin): A lack of healthy RBCs circulating in the blood can cause the skin to appear pale or whitish.
- Jaundice (yellowing of the skin and eyes): When red blood cells break down, they release a yellow pigment called bilirubin. If the body can't eliminate bilirubin fast enough, it builds up in the bloodstream, causing the skin and whites of the eyes to turn yellow.
Other Potential Symptoms
- Dark urine (hemoglobinuria): In some cases, free hemoglobin released from destroyed RBCs can be filtered by the kidneys and appear in the urine, causing a reddish or dark brown discoloration.
- Shortness of breath: This can occur as the body struggles to get enough oxygen to tissues, especially during exertion.
- Rapid heart rate (tachycardia): The heart may beat faster to try and compensate for the reduced oxygen-carrying capacity of the blood.
- Enlarged spleen (splenomegaly): The spleen is an organ that filters blood and removes damaged RBCs. In hemolytic anemia, the spleen may become enlarged due to increased workload from excessive RBC destruction.
Complications
Hemolytic anemia, if left untreated or if severe, can lead to various complications. These complications arise from the ongoing destruction of red blood cells (RBCs) and the body's struggle to compensate.
- Gallstones: Increased breakdown of RBCs leads to the production of excess bilirubin. When bilirubin levels are high, it can solidify and form gallstones in the gallbladder.
- Increased risk of infections: A shortage of healthy RBCs can impair the immune system's ability to fight infections effectively.
- Heart problems: Chronic anemia forces the heart to work harder to pump oxygen-deficient blood throughout the body. This can lead to heart enlargement, arrhythmias (irregular heartbeat), and eventually heart failure.
- Delayed growth and development (in children): In children with chronic hemolytic anemia, oxygen deficiency can impede growth and development.
- Splenic complications: An enlarged spleen due to excessive RBC destruction can rupture, although rare.
- Iron overload (hemochromatosis): The body normally recycles iron from broken-down RBCs. However, in chronic hemolytic anemia, excessive iron absorption can occur, leading to a condition called hemochromatosis, which can damage organs like the liver, heart, and pancreas.
Additional complications specific to certain causes of hemolytic anemia
- March hemoglobinuria: This rare condition can lead to kidney damage if severe and prolonged.
- Paroxysmal nocturnal hemoglobinuria (PNH): This rare blood disorder can also cause symptoms like abdominal pain, blood clots, and bone marrow problems.
- Sickle cell disease: In sickle cell disease, a specific type of hemolytic anemia, abnormally shaped RBCs can block blood flow, leading to painful leg ulcers.
Laboratory Investigations
Diagnosing hemolytic anemia involves a multi-step approach, combining information from a patient's medical history and physical examination with various laboratory tests.
Medical History and Physical Examination
- Enquire for symptoms like fatigue, weakness, shortness of breath, and any history of jaundice or dark urine.
- Family history of blood disorders is also important, especially for hereditary hemolytic anemia.
- Physical examination will assess for signs like pale skin, jaundice, and an enlarged spleen (splenomegaly).
The investigation of hemolytic anemia follows a hierarchical approach: first, confirming that anemia is present; second, proving that hemolysis is the cause; and finally, pinpointing the specific underlying mechanism (etiology).
General Screening and Confirmation of Anemia
These tests establish the baseline and confirm the body's attempt to compensate for blood loss.
- Complete Blood Count (CBC): Usually shows decreased Hemoglobin (Hb) and Hematocrit (Hct). The Mean Corpuscular Volume (MCV) is often elevated (macrocytic) because reticulocytes are larger than mature red cells.
- Reticulocyte Count: This is the most critical initial test. A high reticulocyte count (reticulocytosis) indicates the bone marrow is healthy and responding to the premature destruction of RBCs.
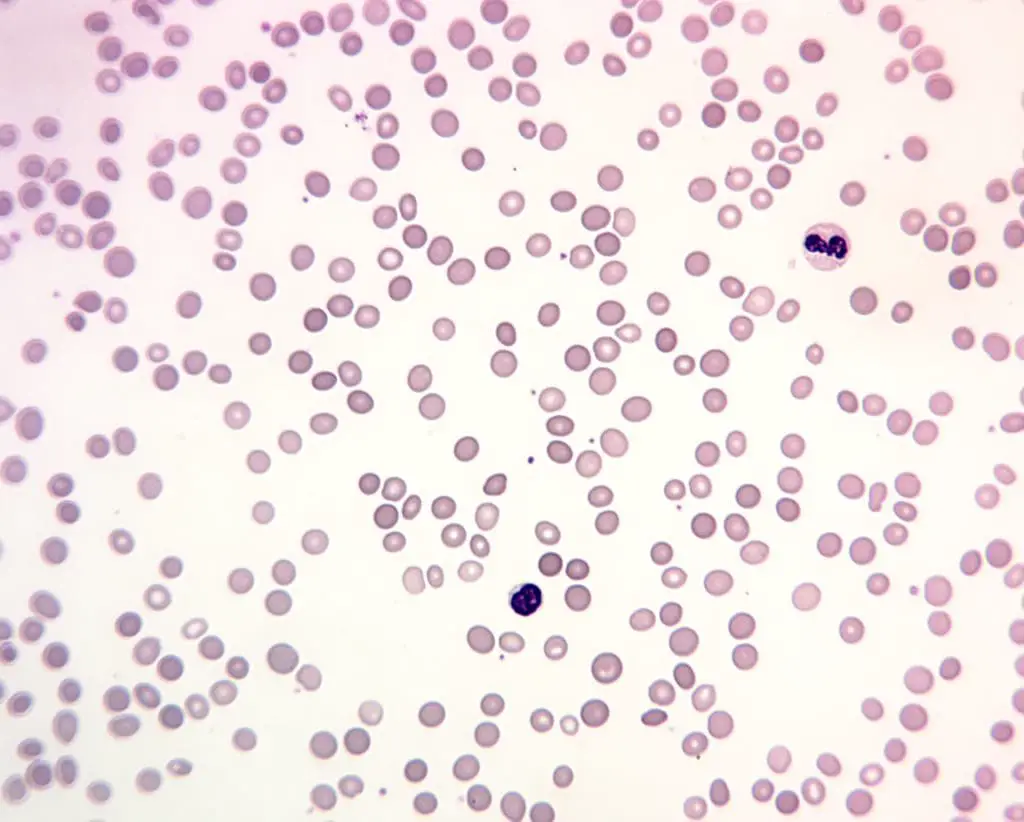
- Peripheral Blood Smear: A manual review of the blood under a microscope to identify "clues" in cell shape:
- Spherocytes: Seen in Hereditary Spherocytosis or Warm Autoimmune Hemolytic Anemia.
- Schistocytes (Fragments): Indicate mechanical destruction (e.g., TTP, HUS, or prosthetic valves).
- Bite Cells/Blister Cells: Suggest oxidative stress, commonly seen in G6PD deficiency.
- Sickle Cells: Diagnostic for Sickle Cell Disease.
Evidence of Active Hemolysis
These markers detect the "byproducts" released when red blood cells rupture.
| Test | Typical Result | Clinical Significance |
| Serum LDH | Increased | Lactate dehydrogenase is an intracellular enzyme; its elevation reflects the magnitude of cell turnover. |
| Unconjugated Bilirubin | Increased | Heme breakdown leads to indirect (unconjugated) bilirubin, often causing clinical jaundice. |
| Serum Haptoglobin | Decreased | This protein "mops up" free hemoglobin. Low levels are a very sensitive marker for hemolysis. |
| Plasma Free Hb | Increased | Highly suggestive of intravascular hemolysis where RBCs burst in the stream. |
| Urine Hemosiderin | Positive | Found in chronic intravascular hemolysis as iron deposits in the renal tubules are shed. |
Specific Tests for Etiology
Once hemolysis is confirmed, these tests determine why it is happening.
Immune-Mediated Investigations
- Direct Antiglobulin Test (DAT/Coombs Test): Detects antibodies (IgG) or complement (C3) bound to the surface of RBCs. This is the gold standard for diagnosing Autoimmune Hemolytic Anemia (AIHA).
- Indirect Antiglobulin Test (IAT): Used to detect antibodies in the patient's serum, primarily for cross-matching before transfusions.
Membrane and Enzyme Investigations
- Flow Cytometry (EMA Binding): The modern preferred test for Hereditary Spherocytosis. It measures the fluorescence of a dye that binds to the RBC membrane.
- G6PD Enzyme Assay: Measures the activity of the G6PD enzyme. This test can be falsely normal during an acute hemolytic crisis because the older cells (with the least enzyme) have already been destroyed, leaving only "young" cells with higher enzyme levels.
- Osmotic Fragility Test: An older test where RBCs are placed in increasingly dilute saline. Spherocytes burst more easily than normal cells.
Hemoglobin and Genetic Investigations
- Hemoglobin Electrophoresis / HPLC: Used to identify abnormal hemoglobin variants like HbS (Sickle Cell) or quantify HbA2 and HbF levels to diagnose Thalassemia.
- Next-Generation Sequencing (NGS): Increasingly used for complex or "Coombs-negative" anemias where multiple genetic mutations (membrane proteins + enzymes) are suspected.
Summary of Diagnostic Signatures
- Intravascular Signature: Low haptoglobin + high LDH + hemoglobinuria + schistocytes.
- Extravascular Signature: Normal/low haptoglobin + high bilirubin + splenomegaly + spherocytes.
- Immune Signature: Positive DAT + spherocytes.
Treatment and Management
The management of hemolytic anemia is highly individualized, shifting from emergency stabilization in acute crises to long-term disease modification in chronic hereditary conditions. Treatment strategies are generally divided into three pillars: Supportive Care, Addressing the Underlying Cause, and Novel Targeted Therapies.
General Supportive Care
Regardless of the cause, several supportive measures are universal for patients experiencing high red blood cell turnover.
- Folic Acid Supplementation: Chronic hemolysis leads to "hyperactive" erythropoiesis. To prevent a megaloblastic crisis, patients typically require daily folic acid (5 mg) to support the bone marrow's increased production of new cells.
- Blood Transfusions: Used judiciously for symptomatic anemia (tachycardia, dyspnea) or when hemoglobin levels drop below critical thresholds (usually 7 - 8 g/dL). In chronic cases like Thalassemia, iron chelation therapy must be started early to prevent secondary hemochromatosis.
- Hydration: Especially critical in intravascular hemolysis to protect the kidneys from the toxic effects of free hemoglobin and to prevent acute tubular necrosis.
Management of Acquired (Extrinsic) Hemolytic Anemia
Management here focuses on suppressing the immune system or removing the external "insult" damaging the cells.
| Condition | Primary Management Strategy |
| Warm AIHA (IgG) | Corticosteroids (Prednisone) are first-line. Second-line treatments include Rituximab or Splenectomy. |
| Cold Agglutinin (IgM) | Cold avoidance is paramount. Traditional steroids are often ineffective; Rituximab or newer C1s inhibitors (Sutimlimab) are preferred. |
| MAHA (TTP/HUS) | Plasma Exchange (PEX) is a medical emergency for TTP to remove ADAMTS13 inhibitors. Avoid platelet transfusions unless life-threatening bleeding occurs. |
| Drug-Induced | Immediate withdrawal of the offending agent (e.g., Penicillin, Quinine). |
| Infectious (Malaria) | Specific anti-parasitic therapy (e.g., Artemisinin-based combinations). |
Management of Hereditary (Intrinsic) Hemolytic Anemia
Because these are genetic, management focuses on symptom control and preventing complications.
- G6PD Deficiency: The "treatment" is primarily prevention. Patients must avoid oxidative triggers such as fava beans, naphthalene (mothballs), and certain medications (e.g., Rasburicase, Primaquine, Nitrofurantoin).
- Hereditary Spherocytosis (HS): Splenectomy remains the definitive treatment for moderate-to-severe cases to stop the extravascular destruction of spherocytes. It is typically delayed until after age 6 to reduce the risk of Overwhelming Post-Splenectomy Infection (OPSI).
- Sickle Cell Disease: Management includes Hydroxyurea (to increase HbF), prophylactic antibiotics, and Voxelotor to prevent sickling.
Recent Advances & Targeted Therapies
The landscape of hematology is shifting toward molecular-level interventions that address the specific enzymatic or protein defects.
- Pyruvate Kinase Activators (Mitapivat): For patients with PK deficiency, this oral medication increases the activity of the enzyme, stabilizing the RBC and significantly reducing the need for transfusions.
- Complement Inhibitors:
- Sutimlimab: Targets the C1s protein in the classical complement pathway, specifically for Cold Agglutinin Disease.
- Pegcetacoplan: A C3 inhibitor used in PNH to prevent both intravascular and extravascular hemolysis (which can occur with older C5 inhibitors).
- Luspatercept: A recombinant fusion protein that enhances RBC maturation; it is increasingly used to treat anemia in Beta-Thalassemia patients who are transfusion-dependent.
Surgical and Interventional Options
- Bone Marrow Transplant (HSCT): Currently the only potential cure for severe cases of Sickle Cell Disease or Thalassemia, though it carries significant risks and requires a matched donor.
- Splenectomy: By removing the primary site of extravascular hemolysis, the lifespan of abnormal RBCs is significantly extended. Patients undergoing splenectomy must receive vaccinations against encapsulated organisms (S. pneumoniae, N. meningitidis, H. influenzae) at least 2 weeks prior to the procedure.
- Cholecystectomy: Patients with chronic hemolysis often develop pigment gallstones (bilirubinate stones). If symptomatic, the gallbladder is typically removed, often during the same session as a splenectomy.
Prognosis
The prognosis for hemolytic anemia varies depending on the type and severity of the condition. Early diagnosis and proper management can significantly improve the quality of life and life expectancy for many patients with hemolytic anemia.
Frequently Asked Questions (FAQs)
What deficiency causes hemolytic anemia?
The most common enzyme deficiency that causes hemolytic anemia is glucose-6-phosphate dehydrogenase (G6PD) deficiency.
Who is most at risk for hemolytic anemia?
The risk factors for hemolytic anemia can be categorized based on whether it's hereditary or acquired.
Hereditary Hemolytic Anemia
- Family History: Having a family member with a hereditary hemolytic anemia significantly increases your risk. These conditions are passed down through genes, so if a parent has a specific type, their children have a higher chance of inheriting it.
- Ethnicity: Certain ethnicities are more prone to specific hereditary hemolytic anemia types. For example, sickle cell disease is more prevalent in people of African descent, while thalassemia is more common in people of Mediterranean, Middle Eastern, and Southeast Asian origin.
Acquired Hemolytic Anemia
- Age: While hemolytic anemia can occur at any age, some acquired forms are more frequent in specific age groups. For instance, autoimmune hemolytic anemia can develop at any time, while G6PD deficiency is typically present from birth.
- Underlying Medical Conditions: Certain medical conditions like autoimmune diseases (lupus, HIV), infections (malaria, babesiosis), and some blood cancers can increase the risk of acquiring hemolytic anemia.
- Medications: Certain medications can damage red blood cells or trigger an immune reaction leading to hemolysis. People taking these medications are at higher risk.
- Blood Transfusions: In rare cases, blood transfusions can lead to hemolytic transfusion reactions if incompatible blood types are mixed.
- Splenic dysfunction: A diseased or malfunctioning spleen can sometimes contribute to hemolytic anemia.
- Environmental factors: Exposure to certain toxins or chemicals can damage red blood cells, leading to hemolytic anemia.
Additional Considerations
- Blood Type: People with certain blood types (e.g., Rh-negative) may be more susceptible to hemolytic transfusion reactions.
- Autoimmune diseases: Women are more likely than men to develop autoimmune disorders that can cause autoimmune hemolytic anemia.
- Geographic location: The prevalence of certain infections that can trigger hemolytic anemia can vary depending on geographic location.
Can hemolytic anemia be cured?
Whether hemolytic anemia can be cured depends on the underlying cause:
| Type of Hemolytic Anemia | Can it be Cured? | Explanation |
| Hereditary | No (but manageable) | Caused by genetic mutations, so a true cure is difficult. However, management strategies can significantly improve symptoms. |
| Acquired (Curable Causes) | Yes (depending on cause) | If the cause is an infection, medication, or a temporary condition, addressing it can potentially cure the hemolytic anemia. |
| Acquired (Manageable Causes) | No (but manageable) | In cases like autoimmune hemolytic anemia, a complete cure might not be possible. However, long-term management with medications can control the condition. |
Is thalassemia a hemolytic anemia?
Yes, thalassemia is a type of hemolytic anemia. It's actually one of the most common inherited forms of hemolytic anemia. There are different types of thalassemia based on which globin chain production is affected (alpha or beta) and the severity of the mutation.
What organ is affected by hemolytic anemia?
While no single organ is directly "affected" by hemolytic anemia, several organs are involved in the disease process. The breakdown of red blood cells with increased destruction of RBCs - the spleen has to work harder, and it might enlarge (splenomegaly); the body's response to compensate - the bone marrow typically increases RBC production to compensate for the ongoing destruction; and the consequences of increased cell destruction all have an impact on various organs - with increased RBC breakdown, the liver may become overloaded, leading to elevated bilirubin levels and potentially causing jaundice (yellowing of the skin and eyes).
Is hemolytic anemia painful?
In some cases, hemolytic anemia can be associated with pain:
- Splenomegaly (enlarged spleen): A severely enlarged spleen due to excessive red blood cell destruction can sometimes cause discomfort or a dull ache in the upper left abdomen.
- Gallstones: The breakdown of red blood cells increases the production of bilirubin, which can solidify and form gallstones in the gallbladder. Gallstones can cause sharp pain in the upper right abdomen, especially after eating fatty foods.
- Leg ulcers (sickle cell disease): In sickle cell disease, a specific type of hemolytic anemia, damaged red blood cells can block blood flow, leading to painful ulcers in the legs.
What is the difference between anemia and hemolytic anemia?
Anemia is a general term for a condition where your blood has a lower than normal number of red blood cells (RBCs) or not enough hemoglobin, the protein in RBCs that carries oxygen. This deficiency in healthy RBCs leads to reduced oxygen delivery throughout the body, causing symptoms like fatigue, weakness, and shortness of breath.
Hemolytic anemia is a specific type of anemia where red blood cells are destroyed prematurely. This excessive destruction disrupts the normal lifespan of RBCs, leading to a shortage of healthy oxygen carriers in the bloodstream and causing the symptoms of anemia.
Think of anemia as a broad category, and hemolytic anemia as a specific subtype within that category caused by a particular mechanism (excessive red blood cell destruction).
Is hemolytic anemia a form of cancer?
While some cancers, particularly cancers of the blood and bone marrow (like leukemia), can cause hemolytic anemia as a complication, hemolytic anemia itself is not a form of cancer. It's a separate blood disorder with distinct causes and mechanisms.
Can hemolytic anemia cause death?
Hemolytic anemia itself is rarely fatal, especially if diagnosed early and treated properly. However, there are some situations where it can lead to serious complications that increase the risk of death.
Complications that can be life-threatening
- Severe anemia: Extremely low red blood cell count can significantly impair oxygen delivery to vital organs like the heart and brain. This can lead to heart failure, stroke, or coma in severe cases.
- Heart problems: Chronic hemolytic anemia forces the heart to work harder to pump enough oxygen-rich blood throughout the body. This can lead to heart enlargement (cardiomegaly) and eventually heart failure.
- Splenic rupture (rare): A severely enlarged spleen due to excessive red blood cell destruction can rarely rupture, causing internal bleeding and requiring emergency medical attention.
- Increased risk of infections: A chronic shortage of red blood cells can weaken the immune system, making you more susceptible to serious infections that can be life-threatening.
Additionally, the underlying cause of hemolytic anemia can also contribute to the risk of death. For example, severe cases of malaria or babesiosis (parasitic infections) that cause hemolytic anemia can be fatal if not treated promptly.
What is the life expectancy of a person with hemolytic anemia?
Hereditary hemolytic anemia: With proper management, some individuals with hereditary hemolytic anemia can lead full lifespans or close to it. However, conditions like severe sickle cell disease can have a significant impact on life expectancy.
Acquired hemolytic anemia: The life expectancy for acquired hemolytic anemia depends heavily on the underlying cause. If the cause is treatable or manageable, the life expectancy might not be significantly affected.
Can hemolytic anemia cause liver damage?
Hemolytic anemia doesn't directly damage the liver, but it can cause a buildup of bilirubin and potentially contribute to complications like jaundice and gallstones. The severity of the impact on the liver depends on the degree of hemolytic anemia and the liver's health beforehand.
Can diet or specific foods trigger hemolytic anemia?
In most types of hemolytic anemia, diet does not directly trigger red blood cell destruction. However, individuals with an inherited enzyme defect called Glucose-6-Phosphate Dehydrogenase (G6PD) deficiency can experience severe acute hemolytic episodes after consuming fava beans (a condition known as favism).
What is the difference between warm and cold autoimmune hemolytic anemia?
The difference lies in the optimal temperature at which the autoantibodies bind to red blood cells. In Warm Autoimmune Hemolytic Anemia (WAIHA), IgG antibodies bind to RBCs at normal body temperature (around 37°C), leading to destruction primarily in the spleen. In Cold Agglutinin Disease (CAD), IgM antibodies bind to RBCs at lower temperatures (typically in the extremities), triggering the complement system and causing destruction in the liver or directly in the bloodstream.
How does hemolytic anemia impact pregnancy?
Pregnancy naturally increases the body's demand for red blood cells and folic acid. Pre-existing hemolytic anemia can worsen during pregnancy, increasing the risk of severe maternal anemia, fetal growth restriction, or premature birth. Conditions like autoimmune hemolytic anemia may also require careful management of immunosuppressive medications to ensure fetal safety.
Is an enlarged spleen (splenomegaly) always present in hemolytic anemia?
No. While splenomegaly is a classic physical finding in extravascular hemolysis (such as hereditary spherocytosis or warm autoimmune hemolytic anemia) due to the spleen working overtime to clear damaged RBCs, it is typically absent in pure intravascular hemolysis, where red blood cells are destroyed directly within the blood vessels rather than in the splenic tissue.
How do we know if the treatment is working?
Success is measured not just by a rising Hemoglobin level, but by a falling Reticulocyte count and stabilizing LDH/Bilirubin levels, indicating that the bone marrow is no longer in "overdrive" and cell destruction has slowed.
Glossary of Related Medical Terms
- Anisopoikilocytosis: A medical term indicating significant variation in both the size (anisocytosis) and shape (poikilocytosis) of red blood cells on a peripheral smear.
- Erythropoiesis: The physiological process of producing new red blood cells within the bone marrow.
- Haptoglobin: A plasma protein produced by the liver that binds free hemoglobin released into the bloodstream, preventing oxidative damage to the kidneys.
- Hemoglobinuria: The presence of free hemoglobin in the urine, often resulting in a dark "cola" or tea color; a hallmark of severe intravascular hemolysis.
- Hemosiderinuria: The shedding of hemosiderin (an iron-storage complex) into the urine, which typically indicates chronic intravascular hemolysis.
- Reticulocytosis: An increase in immature red blood cells (reticulocytes) in the peripheral blood, representing the bone marrow's compensatory response to red blood cell destruction.
- Schistocytes: Fragmented, helmet-shaped red blood cells resulting from mechanical shearing in small, damaged blood vessels; highly characteristic of microangiopathic hemolytic anemias (MAHA).
- Spherocytes: Abnormally round, dense red blood cells that lack a central area of pallor. Their rigid structure makes them highly vulnerable to destruction by macrophages in the spleen.
Disclaimer: This article is intended for informational purposes only and is specifically targeted towards medical students. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Baldwin C, Pandey J, Olarewaju O. Hemolytic Anemia. [Updated 2023 Jul 24]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-.
- https://www.msdmanuals.com/professional/hematology-and-oncology/anemias-caused-by-hemolysis/autoimmune-hemolytic-anemia
- Phillips J, Henderson AC. Hemolytic Anemia: Evaluation and Differential Diagnosis. Am Fam Physician. 2018 Sep 15;98(6):354-361. PMID: 30215915.
- Goldberg S, Hoffman J. Clinical Hematology Made Ridiculously Simple, 1st Edition: An Incredibly Easy Way to Learn for Medical, Nursing, PA Students, and General Practitioners (MedMaster Medical Books). 2021.
- Keohane EM, Otto CN, Walenga JM. Rodak's Hematology 6th Edition (Saunders). 2019.
- Kubo, A., Murakami, S., & Iwata, T. (2024). Drug Interaction-induced Hemolytic Anemia: An Unresolved Diagnostic Process. Internal medicine (Tokyo, Japan), 63(5), 631–633. https://doi.org/10.2169/internalmedicine.2119-23
- Zantek, N. D., Koepsell, S. A., Tharp, D. R., Jr, & Cohn, C. S. (2012). The direct antiglobulin test: a critical step in the evaluation of hemolysis. American journal of hematology, 87(7), 707–709. https://doi.org/10.1002/ajh.23218
- Barcellini, W., & Fattizzo, B. (2015). Clinical Applications of Hemolytic Markers in the Differential Diagnosis and Management of Hemolytic Anemia. Disease markers, 2015, 635670. https://doi.org/10.1155/2015/635670
- Gertz M. A. (2022). Updates on the Diagnosis and Management of Cold Autoimmune Hemolytic Anemia. Hematology/oncology clinics of North America, 36(2), 341–352. https://doi.org/10.1016/j.hoc.2021.11.001
- Hill, A., & Hill, Q. A. (2018). Autoimmune hemolytic anemia. Hematology. American Society of Hematology. Education Program, 2018(1), 382–389. https://doi.org/10.1182/asheducation-2018.1.382
- Kalfa T. A. (2016). Warm antibody autoimmune hemolytic anemia. Hematology. American Society of Hematology. Education Program, 2016(1), 690–697. https://doi.org/10.1182/asheducation-2016.1.690
- Mohandas N. (2018). Inherited hemolytic anemia: a possessive beginner's guide. Hematology. American Society of Hematology. Education Program, 2018(1), 377–381. https://doi.org/10.1182/asheducation-2018.1.377