Key Takeaways
A red blood cell (RBC, or erythrocyte) is a biconcave, anucleate cell whose central job is to ferry oxygen from the lungs to tissues and carbon dioxide back to the lungs. About 1.9 billion people worldwide live with anemia, making red blood cell physiology one of the most clinically relevant topics in medicine [1].
- Erythropoiesis ▾: Red blood cells are made in the bone marrow through erythropoiesis, a process that takes roughly 7 days from stem cell to circulating cell and is regulated mainly by erythropoietin (EPO), a hormone released from the kidney when tissues sense low oxygen [1,5].
- RBC Metabolism ▾: RBCs rely on pathways like Embden-Meyerhof, Hexose Monophosphate, Methemoglobin Reductase, and Leubering-Rapoport Shunt for energy and to maintain hemoglobin function and cell integrity.
- Red Blood Cell Membrane ▾: A complex and dynamic structure vital for RBC survival and function, protecting the cell, maintaining its biconcave shape (crucial for efficient oxygen transport), and regulating molecular transport. It's composed of a lipid bilayer and various proteins (e.g., spectrin, ankyrin) that form a supporting cytoskeleton.
- Hemoglobin ▾: Hemoglobin inside the RBC carries oxygen using four heme groups. Its sigmoid oxygen-binding curve is shifted by pH (the Bohr effect), CO₂, temperature, and 2,3-DPG, allowing oxygen delivery to match tissue demand [2].
- RBC Lifespan and Destruction ▾: Each RBC survives about 120 days before macrophages in the spleen, liver, and bone marrow recognize it as aged and break it down, recycling iron and converting heme into bilirubin [2,7].
- Clinical Relevance & Disorders ▾: Disorders of red blood cells include anemias (iron deficiency, B12/folate deficiency, hemolytic anemia, sickle cell disease, thalassemia), polycythemias, and membrane defects such as hereditary spherocytosis [1,3].
*Click ▾ for more information
Introduction
The red blood cell is the most abundant cell in the human body, with about 25 trillion circulating in an adult at any moment. Strip it down and it is almost embarrassingly simple: a flexible bag of hemoglobin with no nucleus and no mitochondria. Yet without it, every other cell in the body would suffocate within minutes.
This article walks through how red blood cells are built, how they work, how they age, and what goes wrong when they don't.
Why red blood cells matter?
Red blood cells do four jobs that keep the body alive. First, they transport oxygen. Hemoglobin, the iron-containing protein packed inside each RBC, picks up oxygen in the lungs and releases it in tissues that need it. Second, they help carry carbon dioxide back to the lungs, partly bound to hemoglobin and partly as bicarbonate generated inside the cell. Third, they buffer blood pH; the same chemistry that lets hemoglobin grab and release CO₂ also stabilizes the body's acid-base balance. Fourth, they participate in nitric oxide signaling where RBCs both scavenge and release NO-related molecules, which fine-tunes blood vessel tone in low-oxygen regions [4].
Beyond these core roles, RBCs help the immune system clear circulating immune complexes through a surface receptor called CR1, and they contribute to the redistribution of body heat by virtue of being the most abundant cell in flowing blood.
How Red Blood Cells Are Made
Hematopoiesis
Every blood cell in the body, including every red blood cell, traces its lineage to a single ancestor: the hematopoietic stem cell (HSC). HSCs are remarkable because they can both copy themselves (self-renewal) and turn into any blood lineage (differentiation).
Inside the bone marrow, an HSC first commits to one of two paths: the myeloid lineage or the lymphoid lineage. Lymphoid progenitors mature into B and T lymphocytes and natural killer cells. Myeloid progenitors give rise to red blood cells, platelets (via megakaryocytes), and granulocytes such as neutrophils, eosinophils, and basophils, plus monocytes.
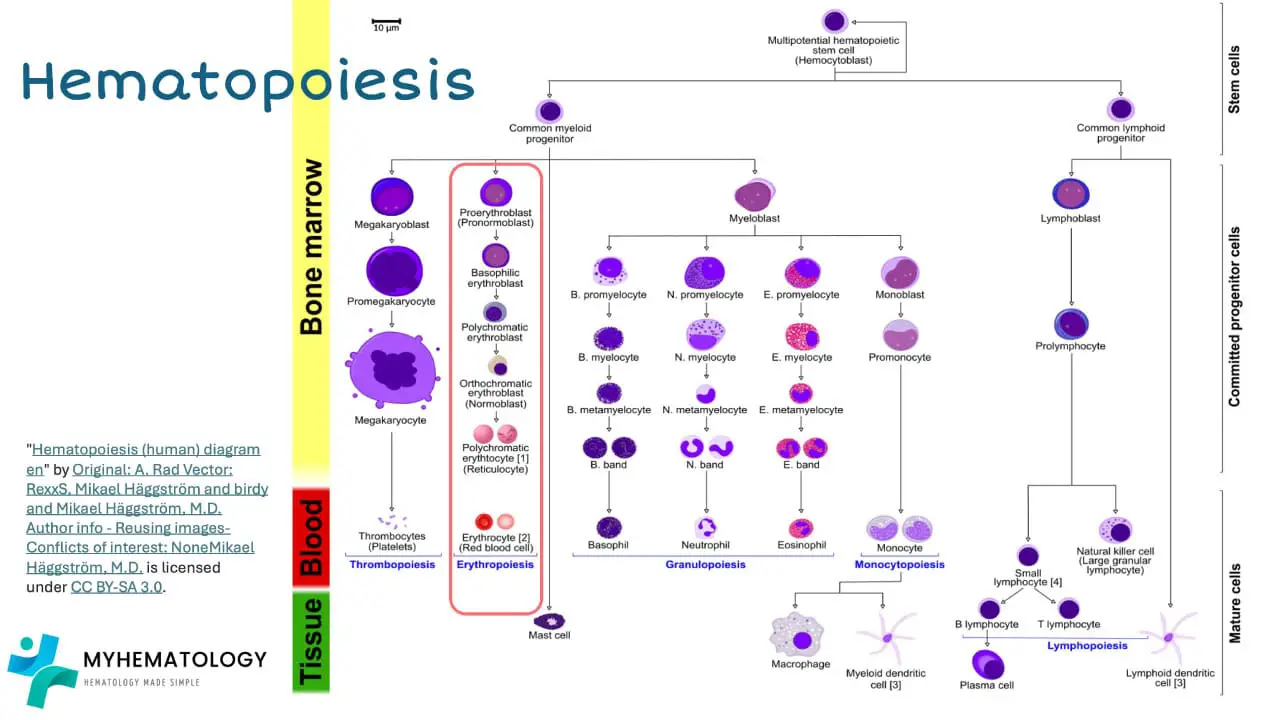
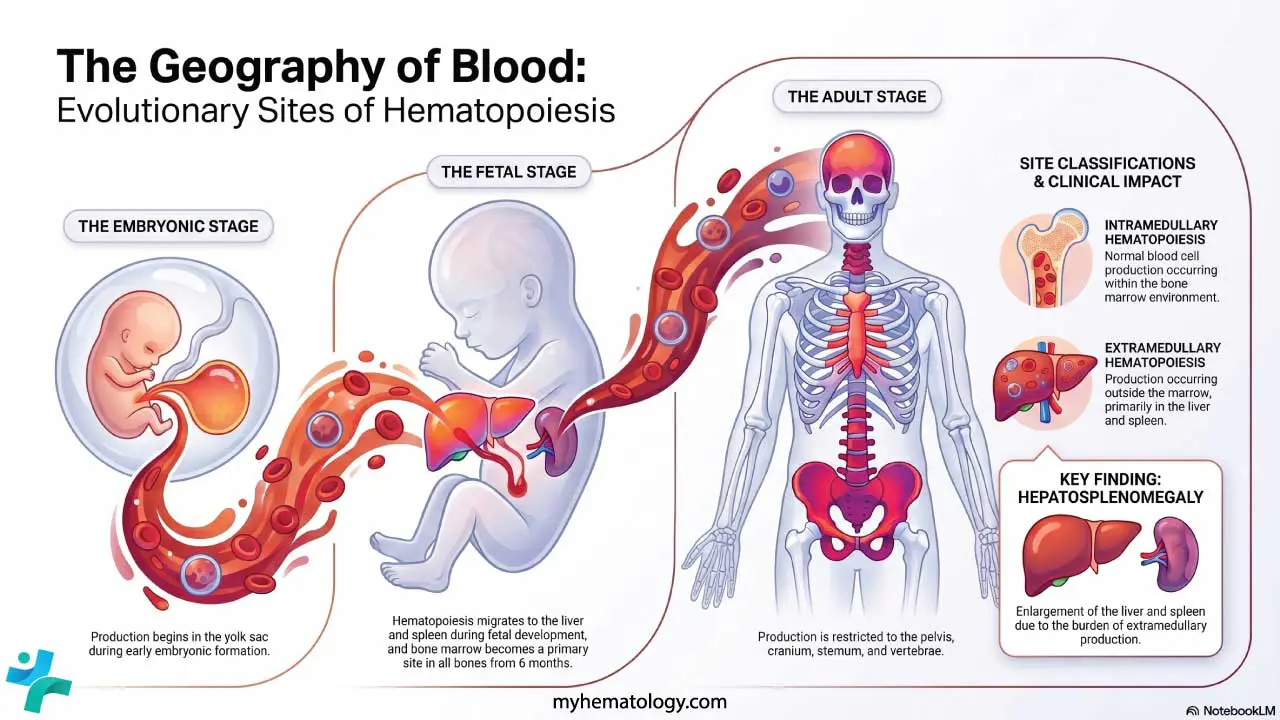
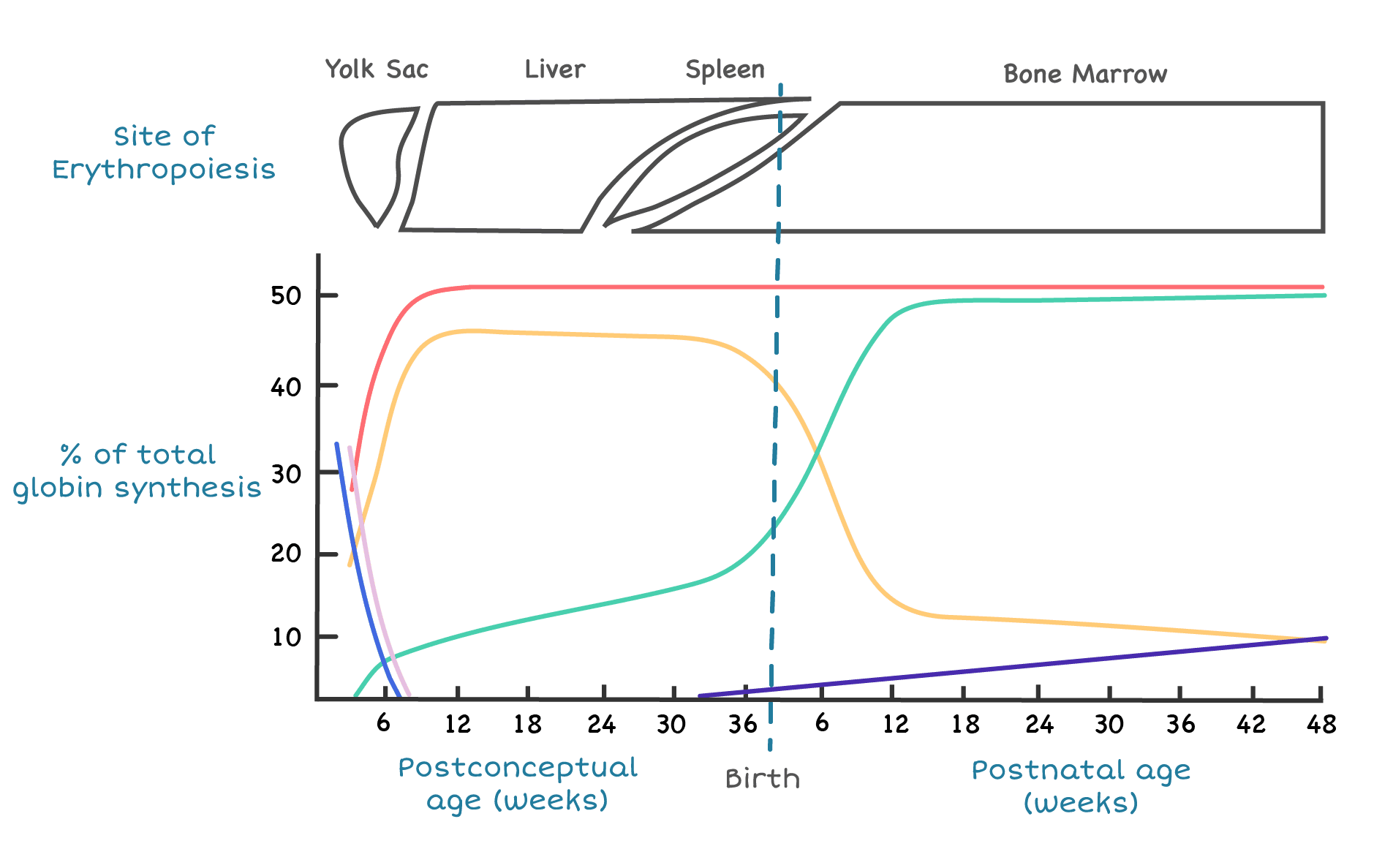
Hematopoiesis sites
In the embryo, blood cell production starts in the yolk sac, then migrates to the liver and spleen during fetal life. From around 6 months of gestation onward, the bone marrow takes over and remains the main site for the rest of life. In children, almost every bone produces blood. By adulthood, active red marrow is concentrated in the pelvis, sternum, vertebrae, ribs, and skull.
When the bone marrow can't keep up for example, in severe hemolytic anemia or marrow infiltration, the liver and spleen can switch their old fetal job back on. This is called extramedullary hematopoiesis, and it physically enlarges those organs (hepatosplenomegaly).
Why the bone marrow microenvironment matters
Hematopoietic stem cells don't float freely; they live in a specialized niche built from stromal cells, osteoblasts, endothelial cells, adipocytes, and macrophages, plus a soup of cytokines and growth factors. This niche keeps HSCs in a resting state most of the time, protects them from damage, and tells them when to divide and what to become. Disrupt the niche and hematopoiesis fails, which is what happens in aplastic anemia and after high-dose chemotherapy.
Erythropoiesis
Erythropoiesis is the specific branch of hematopoiesis that produces red blood cells. The pathway runs:
Hematopoietic stem cell → proerythroblast → early/basophilic erythroblast → polychromatic erythroblast → orthochromatic normoblast → reticulocyte → mature erythrocyte.
As the cell matures, three things happen in parallel: hemoglobin accumulates in the cytoplasm, the nucleus condenses and is eventually extruded, and the cell shrinks. The end product is the small, hemoglobin-packed disk we recognize as an RBC. Reticulocytes still contain residual ribosomal RNA, which is why they stain bluish on a peripheral smear and why the reticulocyte count is a useful index of how hard the marrow is working. In healthy adults, reticulocytes make up roughly 0.5–2% of circulating red cells [2].
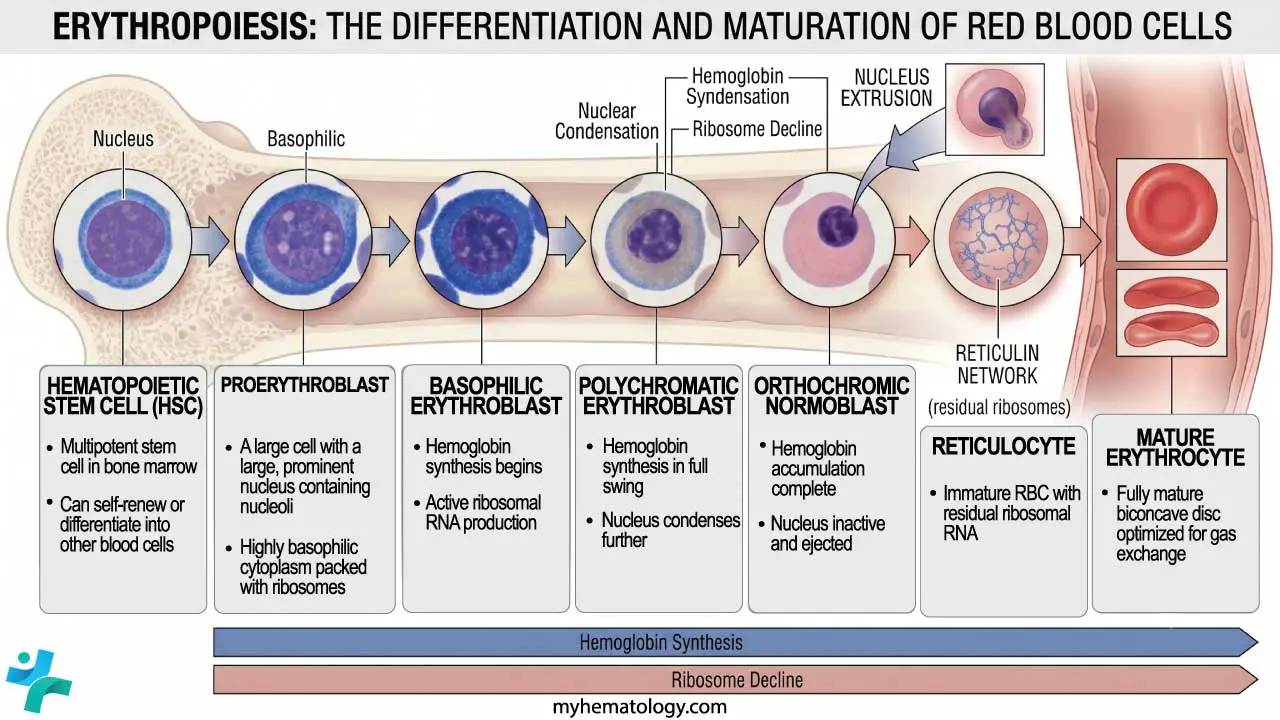
The full marrow journey takes about 7 days, with the last 3–4 days spent as a reticulocyte that is released into circulation and matures fully within 1–2 days.
Regulation of Erythropoiesis
The body adjusts red cell production to oxygen demand through an elegant feedback loop. When tissues sense low oxygen, peritubular interstitial fibroblasts in the kidney cortex (sometimes called REP cells) release erythropoietin (EPO) [1,6]. (Note: many older texts incorrectly attribute EPO production to juxtaglomerular cells; juxtaglomerular cells secrete renin, not EPO.) EPO travels through the blood to the bone marrow, binds receptors on erythroid progenitors, and drives them to proliferate, mature, and avoid apoptosis. The net result is more red cells, more hemoglobin, and more oxygen delivery
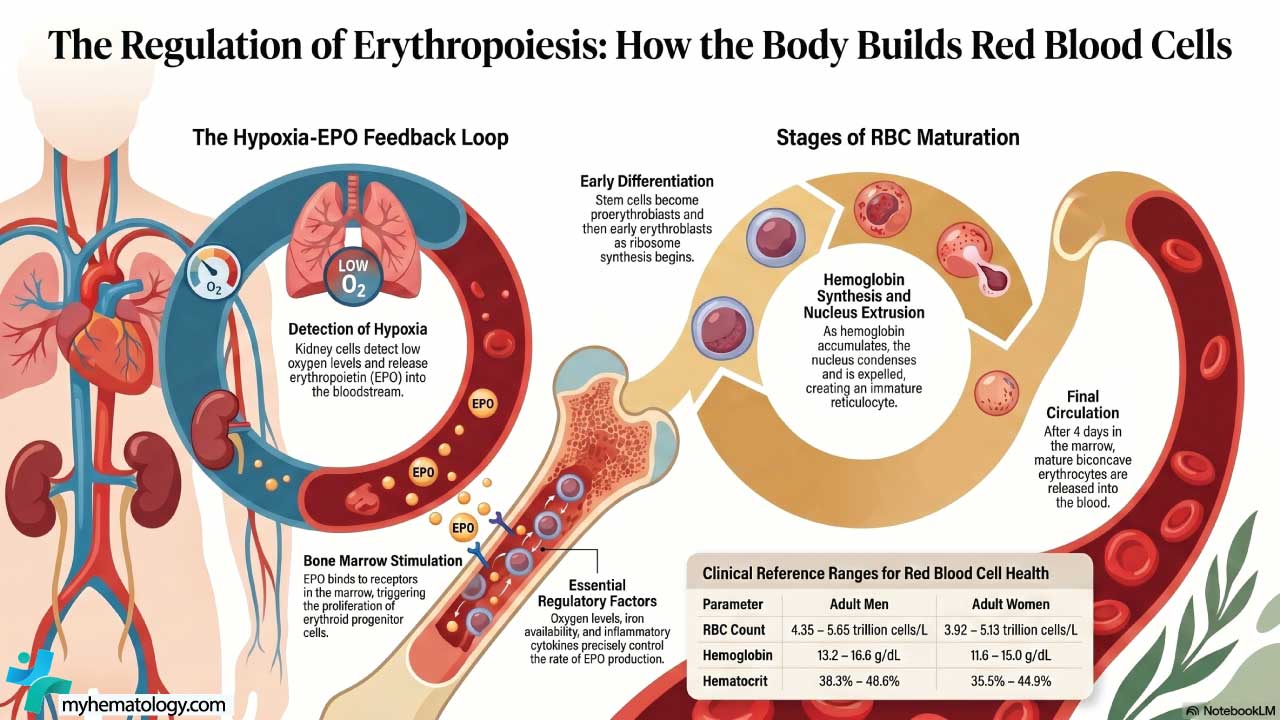
As oxygen levels in the blood decline, specialized cells in the kidneys, known as juxtaglomerular (JAG) cells, detect the hypoxia and release EPO into the bloodstream. EPO travels to the bone marrow, where it binds to receptors on erythroid progenitor cells, initiating a cascade of signaling events that trigger the differentiation and maturation of these cells into red blood cells.
The molecular sensor behind this loop is the hypoxia-inducible factor (HIF) system, recognized with the 2019 Nobel Prize in Physiology or Medicine [5]. In normal oxygen, an enzyme family called prolyl hydroxylases tags HIF-α subunits for destruction. When oxygen drops, those enzymes can't do their job, HIF-2α accumulates, and the EPO gene is switched on. This sensor explains why people at high altitude make more red cells, why kidney failure causes anemia (failed EPO production), and why a new drug class — HIF-prolyl hydroxylase inhibitors such as roxadustat and daprodustat — can treat anemia of chronic kidney disease orally instead of with injected EPO [6].
Characteristics of Red Blood Cells (RBCs)
A red blood cell has no nucleus and no mitochondria. It cannot make new proteins or repair its DNA. Yet it must survive 120 days of being squeezed through capillaries, battered by oxidative stress, and shifted between extremes of pH. Three structural and metabolic features make this possible: a specialized metabolism, a remarkable membrane, and the right kind of hemoglobin.
Red blood cell (RBC) metabolism
Without mitochondria, the RBC depends on glucose and four interlinked cytoplasmic pathways [4].
1. The Embden-Meyerhof pathway (glycolysis)
This is the energy engine. It converts glucose to pyruvate anaerobically and produces about 90–95% of the cell's ATP. ATP powers the membrane ion pumps that keep the cell's shape and volume stable.
The key regulatory enzyme is pyruvate kinase. When this enzyme is deficient (an inherited disorder called PK deficiency), ATP runs short, the cell's membrane fails, and chronic hemolytic anemia follows. Until recently, treatment was supportive — transfusions and, sometimes, splenectomy. Mitapivat, an oral pyruvate kinase activator approved by the FDA in 2022, is the first disease-modifying therapy and is now in trials for thalassemia and sickle cell disease [8].
2. The hexose monophosphate shunt (pentose phosphate pathway)
This pathway generates NADPH, which keeps glutathione in its reduced form. Reduced glutathione is the cell's main antioxidant. Without it, oxidants such as hydrogen peroxide damage hemoglobin and the membrane.
The key enzyme is glucose-6-phosphate dehydrogenase (G6PD). G6PD deficiency is the most common enzymopathy in the world, affecting an estimated 400 million people. Most carriers are well until exposed to a trigger like fava beans, certain antimalarials, sulfa drugs, or infection, at which point oxidative stress overwhelms the cell and acute hemolysis follows.
3. The methemoglobin reductase pathway
Iron in heme exists in the ferrous (Fe²⁺) state, which can carry oxygen. The oxidative environment of the RBC constantly nudges iron toward the ferric (Fe³⁺) state, producing methemoglobin, which cannot carry oxygen. The methemoglobin reductase pathway, mainly using NADH, reduces it back. When this pathway fails, either congenitally or because of oxidizing drugs and chemicals (nitrites, dapsone, benzocaine), methemoglobinemia develops, and the patient looks cyanotic despite a normal PaO₂.
4. The Luebering-Rapoport shunt
This branch of glycolysis produces 2,3-bisphosphoglycerate (2,3-DPG). 2,3-DPG binds to deoxyhemoglobin and lowers its affinity for oxygen, helping unload oxygen in tissues. Levels rise in chronic hypoxia (high altitude, chronic lung disease, anemia) and fall in stored blood, which is why old transfused units initially deliver oxygen poorly.
Red blood cell (RBC) membrane
The RBC is about 7.5 μm across and 2 μm thick, with a biconcave-disc shape that maximizes surface area for gas exchange and, crucially, lets the cell deform. This deformability matters most in the spleen, where RBCs squeeze through interendothelial slits as narrow as 1–3 μm. Cells that can't deform i.e. old, damaged, or membrane-defective get trapped and destroyed there [3].
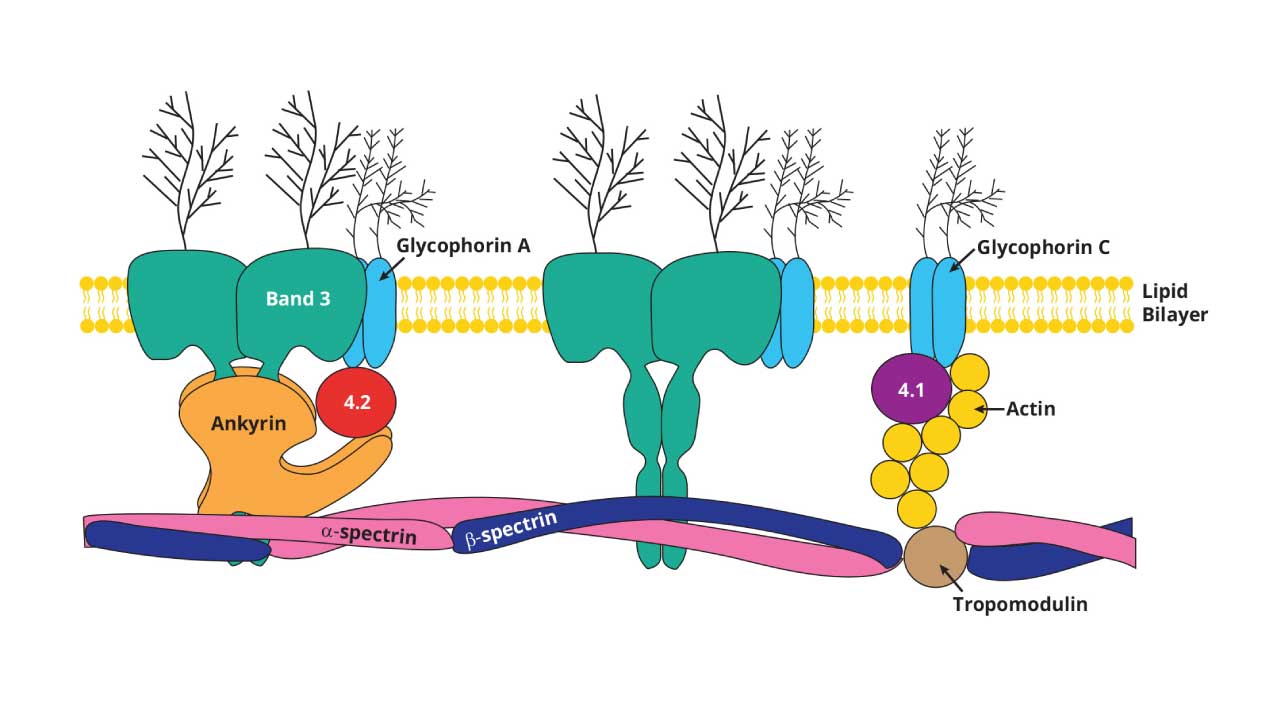
The membrane has three layers of architecture:
- A lipid bilayer with asymmetric phospholipid distribution, embedded with cholesterol and glycolipids that carry blood group antigens (ABO, Rh).
- Integral (transmembrane) proteins, including Band 3 (anion exchange), the glycophorins (which carry MN blood group antigens), and aquaporins (water transport).
- A peripheral cytoskeleton on the inner surface, built mainly from spectrin, actin, ankyrin, protein 4.1, and protein 4.2. This lattice is what gives the membrane its elasticity.
Mutations in any of these scaffolding proteins produce inherited membrane disorders:
- Hereditary spherocytosis — usually defects in ankyrin or spectrin; cells lose surface area and become spherical, then get destroyed in the spleen.
- Hereditary elliptocytosis — spectrin self-association defects produce elliptical cells.
- Hereditary stomatocytosis — cation transport abnormalities give the cell a slit-like central pallor.
Hemoglobin
Hemoglobin makes up roughly 33% of the RBC's weight. Each molecule is a tetramer of four globin chains (two α-like and two β-like), with one heme group nestled inside each chain. Each heme contains one iron atom, and each iron atom binds one oxygen molecule — so one hemoglobin tetramer carries up to four oxygens.
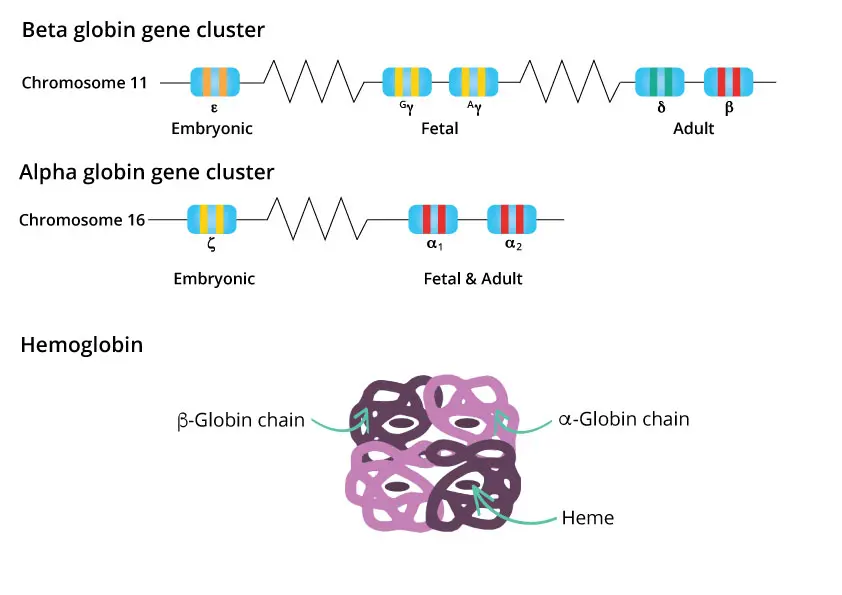
Hemoglobin subtypes change with age
Different globin genes are expressed at different stages of life. The α-like genes sit on chromosome 16; the β-like genes sit on chromosome 11.
- Embryo: Hb Gower-1, Gower-2, Hb Portland.
- Fetus: HbF (α₂γ₂), which has a higher oxygen affinity than HbA so it can pull oxygen across the placenta.
- Adult: about 96–98% HbA (α₂β₂), 2–3% HbA₂ (α₂δ₂), and less than 1% residual HbF.
The fetal-to-adult switch begins before birth and is largely complete by 6 months of age. The molecular master switch is a transcription factor called BCL11A, which silences the γ-globin gene after birth [12]. Disabling BCL11A reactivates HbF, which is exactly how the gene therapy Casgevy (exa-cel) treats sickle cell disease and beta-thalassemia [9].
How hemoglobin releases oxygen: the dissociation curve
The oxygen dissociation curve (ODC) plots the percentage of hemoglobin saturated with oxygen against the partial pressure of oxygen. Its sigmoid (S) shape comes from cooperativity: when one heme binds oxygen, it nudges the other three into a higher-affinity state. This makes hemoglobin a near-ideal courier — it loads up almost completely in the lungs and unloads efficiently in tissues.
The position of the curve shifts with the local environment:
- Right shift (releases oxygen more readily): high CO₂, low pH, high temperature, high 2,3-DPG. This is the Bohr effect, and it is exactly what exercising muscle needs — its acidic, warm, CO₂-rich environment automatically pulls oxygen off hemoglobin.
- Left shift (holds oxygen more tightly): low CO₂, high pH, low temperature, low 2,3-DPG, fetal hemoglobin, carbon monoxide.
Interactive Oxygen Dissociation Curve
Adjust the variables to observe the Bohr effect. The dashed line represents normal physiological baseline.
Clinically, the ODC explains why patients with sepsis, anemia, or chronic lung disease can be oxygenating at the lungs but still oxygen-starved in tissues, and why stored blood needs time to regenerate 2,3-DPG before it delivers oxygen well.
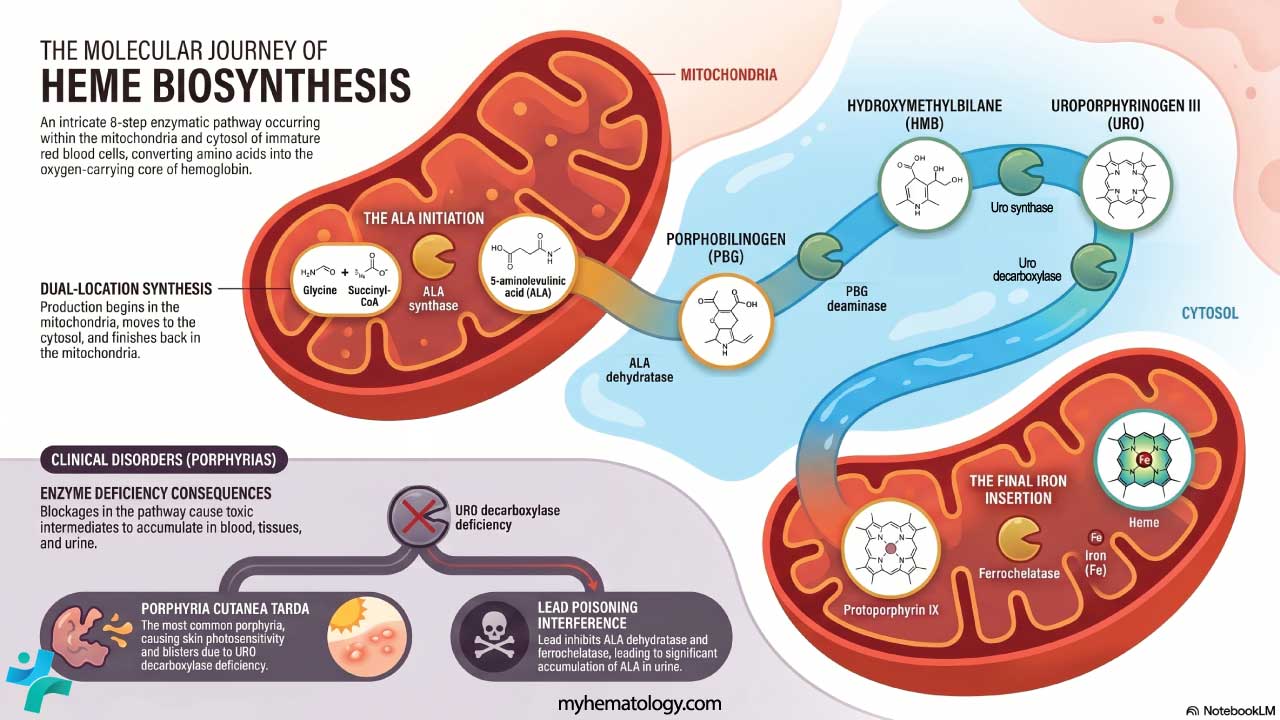
Heme synthesis and the porphyrias
Heme is built in eight enzymatic steps that move between the mitochondria and the cytosol. The pathway starts with glycine and succinyl-CoA (forming aminolevulinic acid, ALA) and ends with ferrochelatase inserting iron into protoporphyrin IX. A defect in any step backs up an intermediate, producing one of the porphyrias — a heterogeneous group of disorders that present with neurovisceral attacks, photosensitivity, or both.
Notable examples include acute intermittent porphyria (HMBS deficiency; severe abdominal pain, neuropsychiatric symptoms, no skin disease), porphyria cutanea tarda (UROD deficiency; the most common form, with photosensitivity and skin blistering), and erythropoietic protoporphyria (ferrochelatase deficiency; painful photosensitivity in childhood). Lead poisoning mimics a porphyria by inhibiting ALA dehydratase and ferrochelatase.
Iron handling and hepcidin
Heme synthesis runs on iron. Body iron is exquisitely controlled by hepcidin, a small peptide hormone made by the liver [7]. Hepcidin blocks ferroportin, the only known iron exporter, so high hepcidin traps iron inside enterocytes and macrophages while low hepcidin releases it into circulation. Hepcidin rises with inflammation (driving the anemia of chronic disease) and falls with iron deficiency and active erythropoiesis. Hepcidin biology is now a drug target as agonists for iron overload and antagonists for inflammatory anemia.
Aging and Destruction of Red Blood Cells
A red blood cell ages because it cannot repair itself. Over 120 days, several things go wrong at once: ATP production falls, the membrane loses cholesterol and phospholipids, oxidative damage accumulates, methemoglobin reductase activity declines, and IgG molecules and senescence markers (such as exposed phosphatidylserine) appear on the surface. Additionally, young red blood cells express high levels of CD47, a transmembrane protein that acts as a "don't eat me" signal by binding to receptors on immune cells. As the cell ages, CD47 is gradually lost or structurally altered. The combination of losing this protective signal and gaining "eat me" markers like phosphatidylserine gives macrophages in the spleen the definitive green light to clear the cell [14].
When that happens, macrophages of the reticuloendothelial system which are mostly in the spleen, with help from the liver and bone marrow, recognize the senescent cell, engulf it, and recycle its parts. About 90% of RBC turnover happens this way; the rest is intravascular hemolysis.
A more recently described mode of RBC death is eryptosis — a programmed, apoptosis-like death triggered by oxidative stress, hyperosmolarity, or energy depletion, marked by phosphatidylserine flipping to the outer membrane leaflet [11]. Eryptosis is now thought to contribute to anemia in sepsis, kidney disease, and metabolic disorders.
Inside the macrophage, hemoglobin is broken into its parts:
- Globin chains are degraded to amino acids and recycled.
- Iron is released, bound to transferrin, and either reused for new hemoglobin or stored in ferritin.
- Heme is converted to biliverdin, then to bilirubin. Bilirubin travels on albumin to the liver, gets conjugated, and is excreted in bile. Gut bacteria convert it to urobilinogen and stercobilin, which color urine yellow and stool brown.
Conditions such as anemia, diabetes, sickle cell disease, and thalassemia accelerate this aging process. Smoking and heavy alcohol use also shorten RBC life.
Clinical Significance of the RBCs
Any drop in oxygen delivery touches every organ. Common symptoms like fatigue, breathlessness, dizziness, pallor, palpitations are usually the first clue. The complete blood count (CBC) with red cell indices (MCV, MCH, MCHC, RDW) is the single most useful starting investigation in hematology.
Anemias
Anemia is defined by low hemoglobin or hematocrit. It is sorted by red cell size:
- Microcytic anemia (low MCV): iron deficiency (most common worldwide), thalassemia, anemia of chronic disease, lead poisoning.
- Macrocytic anemia (high MCV): B12 deficiency, folate deficiency, liver disease, hypothyroidism, alcohol, certain drugs.
- Normocytic anemia (normal MCV): acute blood loss, chronic kidney disease (low EPO), bone marrow failure (aplastic anemia, infiltration), early iron deficiency.
- Hemolytic anemia: premature RBC destruction. Causes include autoimmune hemolytic anemia, sickle cell disease, G6PD deficiency, hereditary spherocytosis, mechanical destruction (microangiopathic hemolytic anemia), and infections such as malaria.
Polycythemia: too many red blood cells
A high red cell count thickens the blood and raises the risk of clotting, stroke, and heart attack. Causes split into:
- Primary: polycythemia vera, a clonal myeloproliferative disorder usually driven by a JAK2 V617F mutation.
- Secondary: a physiological response to true hypoxia (chronic lung disease, cyanotic heart disease, high altitude, sleep apnea) or to inappropriate EPO production from kidney or liver tumors.
- Relative: dehydration concentrates the existing cells without increasing the total mass.
Tests that go beyond the basic CBC
When the CBC suggests an RBC problem, the workup typically expands:
- Peripheral blood smear — the single most informative test in hematology; looks for shape abnormalities (sickle cells, schistocytes, spherocytes, target cells), inclusions, and dysplasia.
- Reticulocyte count and reticulocyte production index — distinguishes "marrow not making cells" from "cells being destroyed."
- Iron studies (ferritin, transferrin saturation) — for microcytic anemia.
- B12, folate — for macrocytic anemia.
- Haptoglobin, LDH, indirect bilirubin, direct antiglobulin test (DAT/Coombs) — for suspected hemolysis.
- Hemoglobin electrophoresis or HPLC — for suspected thalassemia or hemoglobinopathy.
- Bone marrow examination — when peripheral tests don't explain the picture.
Red cell distribution width: a small number with big meaning
RDW measures the variation in RBC size. A high RDW (anisocytosis) suggests a mixed population for example, in early iron deficiency, B12/folate deficiency, or after recent blood loss and brisk reticulocytosis. Beyond hematology, RDW is now an established prognostic marker in heart failure, sepsis, and ICU mortality, even when the patient is not anemic [13].
Transfusion thresholds
Transfusion of packed red blood cells is one of the most common interventions in medicine, and current practice favors a restrictive strategy — transfusing only when truly needed [10].
The 2023 AABB international guidelines recommend, for hospitalized adult patients [10]:
- Hb < 7 g/dL for most stable hospitalized adults.
- Hb < 7.5 g/dL for cardiac surgery patients.
- Hb < 8 g/dL for orthopedic surgery patients and those with pre-existing cardiovascular disease.
- No fixed threshold in active hemorrhage or hemodynamic instability — clinical judgment leads.
Always pair the number with the patient: a young, asymptomatic person with chronic anemia tolerates Hb 7 g/dL well; an elderly patient with coronary disease and chest pain may need transfusion at 8.5 g/dL. Compatibility testing like ABO/RhD typing and crossmatching prevents catastrophic hemolytic transfusion reactions [11].
What's new in red blood cell therapeutics
The last few years have transformed treatment options for several RBC disorders:
- Mitapivat (Pyrukynd / Aqvesme): An oral pyruvate kinase activator. Initially approved in 2022 for PK deficiency, it achieved a major milestone in late 2025 when the FDA approved it for the treatment of anemia in adults with both non-transfusion-dependent and transfusion-dependent alpha- or beta-thalassemia, making it the first oral therapy approved across these forms of adult thalassemia [15].
- Luspatercept (Reblozyl): A TGF-β superfamily ligand trap that reduces ineffective erythropoiesis. Following the COMMANDS trial, its approval was expanded to serve as a first-line treatment for anemia in erythropoiesis-stimulating agent (ESA)-naive adults with lower-risk myelodysplastic syndromes, fundamentally outperforming standard EPO [16]. It is also approved for transfusion-dependent beta-thalassemia.
- HIF-prolyl hydroxylase inhibitors: Oral agents that mimic hypoxia and induce endogenous EPO. While several are approved internationally, cardiovascular safety concerns led the US FDA to reject roxadustat and vadadustat. Only daprodustat (Jesduvroq) secured US FDA approval, and it is strictly indicated for patients with chronic kidney disease who are actively on dialysis [17].
- Gene therapies for hemoglobinopathies: In late 2023, the FDA approved two distinct gene therapies providing functional cures for sickle cell disease. Casgevy (exa-cel) utilizes CRISPR/Cas9 technology to edit the BCL11A enhancer, reactivating fetal hemoglobin. In contrast, Lyfgenia (lovo-cel) is not CRISPR-based; it utilizes a lentiviral vector to insert a modified, functional β-globin gene into the patient's stem cells [9,18].
Frequently Asked Questions (FAQs)
What is a red blood cell and what does it do?
A red blood cell (RBC), also called an erythrocyte, is a small, biconcave-shaped cell that has no nucleus and is packed with hemoglobin. Its main job is to carry oxygen from the lungs to every tissue in the body and bring carbon dioxide back to the lungs to be exhaled. RBCs also help buffer blood pH and contribute to nitric oxide signaling that influences blood flow.
What symptoms suggest dangerously low red blood cells?
Profound fatigue, breathlessness at rest, chest pain, palpitations, dizziness or fainting, marked pallor, cold extremities, confusion, and (in severe cases) signs of shock. A symptomatic patient with stable vital signs may still need urgent transfusion if symptoms reflect tissue hypoxia.
What is a normal red blood cell count?
Reference ranges vary slightly between laboratories, age, sex, and altitude. General adult ranges:
- RBC count: men 4.35–5.65 × 10¹²/L; women 3.92–5.13 × 10¹²/L.
- Hemoglobin: men 13.2–16.6 g/dL; women 11.6–15.0 g/dL.
- Hematocrit: men 38.3–48.6%; women 35.5–44.9%.
- WBC: 3.4–9.6 × 10⁹/L.
- Platelets: 150–400 × 10⁹/L.
What level of red blood cell or hemoglobin is dangerously low?
There is no single number, but rough clinical bands are useful:
- Hb < 6.5 g/dL: life-threatening; transfusion almost always required.
- Hb 6.5–7.9 g/dL: severe anemia; usually symptomatic, often transfused.
- Hb 8–10 g/dL: moderate anemia; treatment depends on cause and symptoms.
What matters most is the rate of the drop, the patient's symptoms, and underlying cardiovascular disease. Acute drops are dangerous at higher numbers than chronic ones.
What causes a low red blood cell count?
A low red blood cell count, called anemia, can come from three broad causes: not making enough RBCs (iron, B12, or folate deficiency; bone marrow failure; chronic kidney disease), losing too many (bleeding), or destroying them too fast (hemolysis from autoimmune disease, sickle cell disease, G6PD deficiency, or infections). The pattern on a complete blood count, especially the MCV and reticulocyte count, points toward the cause.
How long does a red blood cell live, and where does it die?
A healthy RBC circulates for about 120 days. After that, macrophages mostly in the spleen, with some in the liver and bone marrow recognize aging RBCs and break them down. The iron is recycled, the globin chains are reused as amino acids, and the heme ring is converted into bilirubin, which the liver excretes in bile.
What's the difference between hemoglobin and a red blood cell?
A red blood cell is the cell; hemoglobin is the protein inside it. Each RBC contains roughly 270 million hemoglobin molecules, and each hemoglobin molecule has four heme groups that can each carry one oxygen molecule. So the cell is the "container" and hemoglobin is the actual oxygen-binding cargo system.
Why does the kidney control red blood cell production?
Because the kidney is the body's best oxygen sensor. Oxygen delivery to the kidney is roughly proportional to circulating red cell mass, so the kidney can detect anemia or hypoxia early. When it does, peritubular fibroblasts release EPO, which drives the bone marrow to make more red cells. This is why patients with chronic kidney disease almost always become anemic — the sensor and the factory are no longer connected.
What does erythropoietin do, and where is it made?
Erythropoietin (EPO) is a hormone that tells the bone marrow to produce more red blood cells. About 90% of EPO is made by specialized fibroblast-like cells in the kidney cortex (not by juxtaglomerular cells), with a smaller amount made by the liver. When tissues sense low oxygen, a transcription factor called HIF-2α stabilizes and switches on the EPO gene.
Are there cures for inherited red blood cell disorders?
Some, increasingly. Allogeneic stem cell transplant has long offered a cure for sickle cell disease, thalassemia, and other inherited RBC disorders, but requires a matched donor and carries significant risk. As of 2023, two CRISPR-based gene therapies (Casgevy and Lyfgenia) provide a functional cure for sickle cell disease and transfusion-dependent beta-thalassemia using the patient's own edited stem cells. Mitapivat is a disease-modifying drug for pyruvate kinase deficiency [8,9].
How does smoking or living at high altitude change the red blood cell count?
Both lower the oxygen reaching tissues and so trigger the EPO/HIF pathway to make more red cells. Long-term high-altitude residents and chronic heavy smokers often have hematocrits well above sea-level norms. This is secondary polycythemia and it is generally adaptive, though very high hematocrits can raise blood viscosity and clot risk.
Are there new treatments for red blood cell disorders?
Yes, several. Mitapivat (an oral pyruvate kinase activator) is now approved for pyruvate kinase deficiency. HIF-prolyl hydroxylase inhibitors such as roxadustat and daprodustat treat anemia of chronic kidney disease without injected EPO. Casgevy and Lyfgenia, approved in late 2023, are gene therapies that can functionally cure sickle cell disease and transfusion-dependent beta-thalassemia. Luspatercept improves anemia in beta-thalassemia and lower-risk myelodysplastic syndrome.
Glossary of Related Medical Terms
- Erythrocyte — Mature red blood cell. The terms "RBC" and "erythrocyte" mean the same thing.
- Erythropoiesis — The process of making red blood cells, mostly inside the bone marrow.
- Hematopoiesis — The bigger process of making all blood cells (red, white, platelets) from a single stem cell.
- Hemoglobin (Hb) — The iron-containing protein inside RBCs that carries oxygen and carbon dioxide.
- Heme — The iron-containing ring within hemoglobin where oxygen actually binds.
- Reticulocyte — A young, almost-mature RBC that has just left the bone marrow; counting them tells you how hard the marrow is working.
- Erythropoietin (EPO) — A hormone, mostly from the kidneys, that tells the bone marrow to make more RBCs when oxygen is low.
- Hypoxia — Low oxygen in the tissues.
- HIF (Hypoxia-Inducible Factor) — The transcription factor that switches on EPO and other oxygen-response genes when hypoxia is detected.
- Hepcidin — The hormone that controls how much iron enters the bloodstream from the gut and from recycled RBCs.
- Bohr effect — The principle that more CO₂ or acid makes hemoglobin release oxygen more easily.
- Cooperativity — Hemoglobin's ability to bind oxygen more eagerly once the first oxygen molecule is bound, producing the S-shaped (sigmoid) ODC.
- 2,3-DPG (2,3-bisphosphoglycerate) — A small molecule inside RBCs that helps hemoglobin release oxygen to tissues.
- Anemia — Low hemoglobin or low RBC count, leading to reduced oxygen delivery.
- Polycythemia — Too many RBCs.
- Hemolysis — Premature destruction of RBCs.
- Eryptosis — A programmed, suicide-like death of RBCs analogous to apoptosis in nucleated cells.
- Reticuloendothelial system (RES) — Tissue macrophages (mainly in spleen, liver, bone marrow) that clear old RBCs.
- MCV, MCH, MCHC, RDW — Red cell indices on a CBC: MCV is average size, MCH and MCHC describe hemoglobin content, RDW measures size variation.
Disclaimer: This article is intended for educational and informational purposes only. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Bain, B.J., Bates I., Laffan M. A. (2016). Dacie and Lewis practical haematology. Elsevier.
- Mohandas, N., & Gallagher, P. G. (2008). Red cell membrane: past, present, and future. Blood, 112(10), 3939–3948. https://doi.org/10.1182/blood-2008-07-161166
- D'Alessandro, A., Anastasiadi, A. T., Tzounakas, V. L., Nemkov, T., Reisz, J. A., Kriebardis, A. G., Zimring, J. C., Spitalnik, S. L., & Busch, M. P. (2023). Red Blood Cell Metabolism In Vivo and In Vitro. Metabolites, 13(7), 793. https://doi.org/10.3390/metabo13070793
- Gregg L. Semenza. 2020. The Genomics and Genetics of Oxygen Homeostasis. Annual Review Genomics and Human Genetics. 21:183-204. https://doi.org/10.1146/annurev-genom-111119-073356
- Koury, M. J., & Haase, V. H. (2015). Anaemia in kidney disease: harnessing hypoxia responses for therapy. Nature reviews. Nephrology, 11(7), 394–410. https://doi.org/10.1038/nrneph.2015.82
- Ganz T. (2013). Systemic iron homeostasis. Physiological reviews, 93(4), 1721–1741. https://doi.org/10.1152/physrev.00008.2013
- Grace, R. F., Rose, C., Layton, D. M., Galactéros, F., Barcellini, W., Morton, D. H., van Beers, E. J., Yaish, H., Ravindranath, Y., Kuo, K. H. M., Sheth, S., Kwiatkowski, J. L., Barbier, A. J., Bodie, S., Silver, B., Hua, L., Kung, C., Hawkins, P., Jouvin, M. H., Bowden, C., … Glader, B. (2019). Safety and Efficacy of Mitapivat in Pyruvate Kinase Deficiency. The New England journal of medicine, 381(10), 933–944. https://doi.org/10.1056/NEJMoa1902678
- Frangoul, H., Altshuler, D., Cappellini, M. D., Chen, Y. S., Domm, J., Eustace, B. K., Foell, J., de la Fuente, J., Grupp, S., Handgretinger, R., Ho, T. W., Kattamis, A., Kernytsky, A., Lekstrom-Himes, J., Li, A. M., Locatelli, F., Mapara, M. Y., de Montalembert, M., Rondelli, D., Sharma, A., … Corbacioglu, S. (2021). CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. The New England journal of medicine, 384(3), 252–260. https://doi.org/10.1056/NEJMoa2031054
- Carson, J. L., Stanworth, S. J., Guyatt, G., Valentine, S., Dennis, J., Bakhtary, S., Cohn, C. S., Dubon, A., Grossman, B. J., Gupta, G. K., Hess, A. S., Jacobson, J. L., Kaplan, L. J., Lin, Y., Metcalf, R. A., Murphy, C. H., Pavenski, K., Prochaska, M. T., Raval, J. S., Salazar, E., … Pagano, M. B. (2023). Red Blood Cell Transfusion: 2023 AABB International Guidelines. JAMA, 330(19), 1892–1902. https://doi.org/10.1001/jama.2023.12914
- Lang, E., & Lang, F. (2015). Triggers, inhibitors, mechanisms, and significance of eryptosis: the suicidal erythrocyte death. BioMed research international, 2015, 513518. https://doi.org/10.1155/2015/513518
- Sankaran, V. G., & Orkin, S. H. (2013). The switch from fetal to adult hemoglobin. Cold Spring Harbor perspectives in medicine, 3(1), a011643. https://doi.org/10.1101/cshperspect.a011643
- Salvagno, G. L., Sanchis-Gomar, F., Picanza, A., & Lippi, G. (2015). Red blood cell distribution width: A simple parameter with multiple clinical applications. Critical reviews in clinical laboratory sciences, 52(2), 86–105. https://doi.org/10.3109/10408363.2014.992064
- Burger, P., de Korte, D., van den Berg, T. K., & van Bruggen, R. (2012). CD47 in Erythrocyte Ageing and Clearance - the Dutch Point of View. Transfusion medicine and hemotherapy : offizielles Organ der Deutschen Gesellschaft fur Transfusionsmedizin und Immunhamatologie, 39(5), 348–352. https://doi.org/10.1159/000342231
- Taher, A. T., Al-Samkari, H., Aydinok, Y., Besser, M., Boscoe, A. N., Dahlin, J. L., De Luna, G., Estepp, J. H., Gheuens, S., Gilroy, K. S., Glenthøj, A., Sim Goh, A., Iyer, V., Kattamis, A., Loggetto, S. R., Morris, S., Musallam, K. M., Osman, K., Ricchi, P., Salido-Fiérrez, E., … ENERGIZE investigators (2025). Mitapivat in adults with non-transfusion-dependent α-thalassaemia or β-thalassaemia (ENERGIZE): a phase 3, international, randomised, double-blind, placebo-controlled trial. Lancet (London, England), 406(10498), 33–42. https://doi.org/10.1016/S0140-6736(25)00635-X
- Platzbecker, U., Della Porta, M. G., Santini, V., Zeidan, A. M., Komrokji, R. S., Shortt, J., Valcarcel, D., Jonasova, A., Dimicoli-Salazar, S., Tiong, I. S., Lin, C. C., Li, J., Zhang, J., Giuseppi, A. C., Kreitz, S., Pozharskaya, V., Keeperman, K. L., Rose, S., Shetty, J. K., Hayati, S., … Garcia-Manero, G. (2023). Efficacy and safety of luspatercept versus epoetin alfa in erythropoiesis-stimulating agent-naive, transfusion-dependent, lower-risk myelodysplastic syndromes (COMMANDS): interim analysis of a phase 3, open-label, randomised controlled trial. Lancet (London, England), 402(10399), 373–385. https://doi.org/10.1016/S0140-6736(23)00874-7
- Dhillon S. (2020). Daprodustat: First Approval. Drugs, 80(14), 1491–1497. https://doi.org/10.1007/s40265-020-01384-y
- Kanter, J., Walters, M. C., Krishnamurti, L., Mapara, M. Y., Kwiatkowski, J. L., Rifkin-Zenenberg, S., Aygun, B., Kasow, K. A., Pierciey, F. J., Jr, Bonner, M., Miller, A., Zhang, X., Lynch, J., Kim, D., Ribeil, J. A., Asmal, M., Goyal, S., Thompson, A. A., & Tisdale, J. F. (2022). Biologic and Clinical Efficacy of LentiGlobin for Sickle Cell Disease. The New England journal of medicine, 386(7), 617–628. https://doi.org/10.1056/NEJMoa2117175