Procedure-At-A-Glance
Flow cytometry analyzes thousands of individual blood cells per second by passing them through a laser and reading scattered and fluorescent light. Immunophenotyping by flow cytometry identifies cells by the proteins (antigens) they carry on their surface or inside, using antibodies tagged with fluorescent dyes. It is the cornerstone test for diagnosing and classifying leukemias and lymphomas, screening for paroxysmal nocturnal hemoglobinuria, and detecting measurable residual disease (MRD) after treatment.
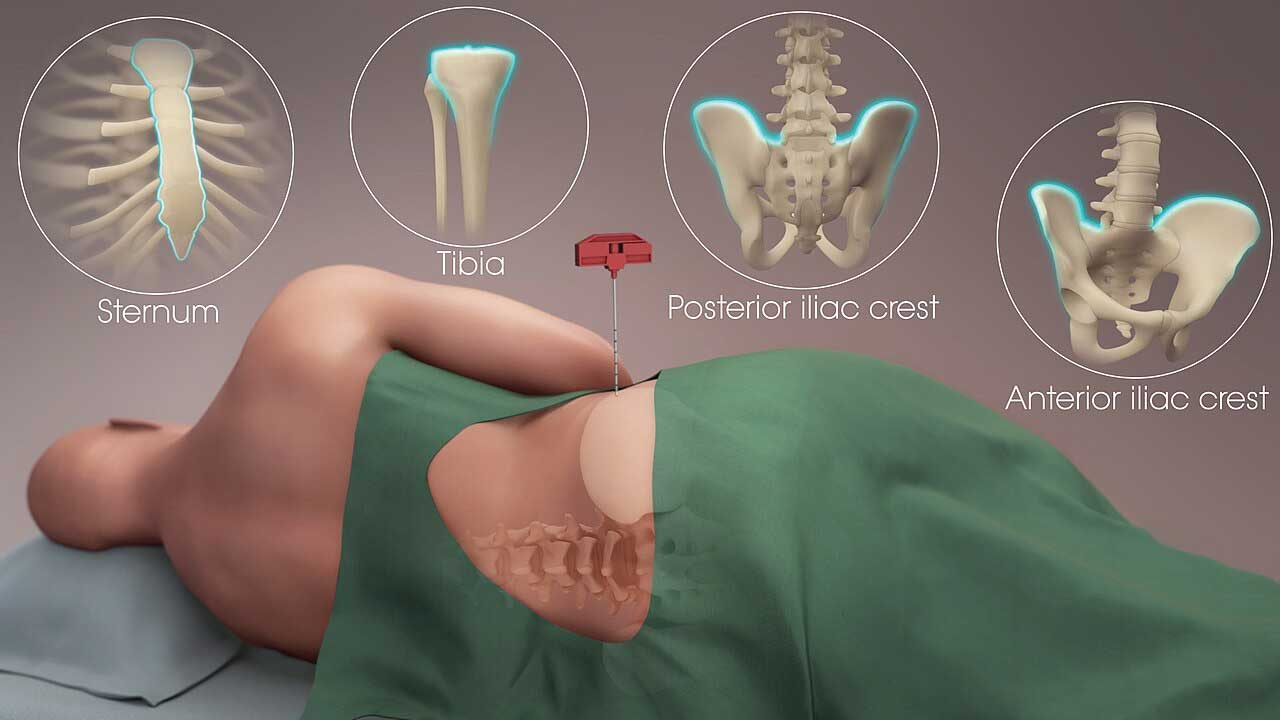
- Sample collection. Whole blood in EDTA (preferred for routine immunophenotyping) or sodium heparin.
- Cell preparation. Either stain-lyse-wash on whole blood (clinical default) or PBMC isolation by density gradient (research default).
- Wash. Resuspend in flow cytometry staining buffer (PBS with BSA or FBS); centrifuge at 250 × g.
- Viability staining. Add a viability dye such as 7-AAD or a fixable amine-reactive dye; incubate in the dark.
- Surface staining. Add fluorochrome-labeled antibodies against surface markers; incubate.
- Optional fixation and permeabilization. Required only for intracellular markers.
- Optional intracellular staining. Add antibodies targeting intracellular antigens.
- Final wash. Remove unbound antibody.
- Acquisition and analysis. Run on the cytometer; apply compensation, gating, and panel-specific analysis.
Introduction
Flow cytometry is the workhorse of modern hematology. In the time it takes to read this paragraph, a clinical flow cytometer can profile tens of thousands of individual blood cells and tell you which are T cells, which are B cells, and which look abnormal enough to suggest leukemia. It is the test behind most modern blood cancer diagnoses [1].
Immunophenotyping is the application of flow cytometry that identifies and characterizes cells by the proteins they express. These proteins, called antigens, act as molecular fingerprints. By staining cells with antibodies tagged to fluorescent dyes (fluorochromes), we can:
- Tell different leukocytes apart — T cells, B cells, NK cells, monocytes, granulocytes.
- Diagnose blood cancers like leukemia and lymphoma by spotting abnormal antigen combinations.
- Monitor how well a patient is responding to treatment.
- Study how the immune system functions in health and disease.
This article walks through both the principles and a working protocol, with a focus on what an undergraduate student or informed caregiver needs to grasp first.
Principle of Flow Cytometry
Flow cytometry combines fluid dynamics, optics, and electronics to interrogate one cell at a time.
1. Single-cell streaming. Cells suspended in saline are pushed through a narrow channel under hydrodynamic focusing, which lines them up single file. This isolation is what makes the technique "single-cell."
2. Laser interrogation. Each cell crosses one or more lasers. The laser interacts with the cell in two ways:
- Scatter. The cell scatters light forward (forward scatter, FSC) and to the side (side scatter, SSC). FSC roughly tracks cell size; SSC roughly tracks internal complexity, which is high in granulocytes and low in lymphocytes.
- Fluorescence. Antibody-bound fluorochromes on or inside the cell absorb laser light and re-emit at characteristic wavelengths, which are caught by detectors.
3. Signal detection. Photomultiplier tubes (PMTs) or, in newer systems, avalanche photodiodes convert light into electrical signals. Each detector is tuned to a wavelength range, allowing several colors to be measured at once.
4. Data analysis. Specialized software plots these signals. For traditional 8- to 12-color panels, analysts manually draw boundaries (gates) around cell populations based on size, complexity, and marker expression using platforms like FlowJo and Infinicyt. However, the complexity of modern high-dimensional data requires advanced approaches. Modern workflows increasingly rely on dimensionality reduction algorithms (such as t-SNE and UMAP) and automated clustering tools (like FlowSOM) to visualize and identify cell populations without manual bias [10].
Conventional vs Spectral Flow Cytometry
Two flavors of flow cytometry now coexist.
Conventional flow cytometry uses optical filters to direct each fluorochrome's emission peak to a dedicated detector. Typical clinical panels carry 8 to 12 colors. Compensation must be set carefully.
Spectral flow cytometry captures the entire emission spectrum of every cell across many detectors and unmixes the signals computationally. This allows panels of up to 40 to 50 markers in a single tube, enabling deep immune profiling and ultra-sensitive rare-event detection from minimal blood volumes [6,11]. Spectral systems are particularly valuable for small samples (pediatric bone marrow, post-chemotherapy specimens) and for detecting rare or subtle abnormal populations.
Both approaches share the same staining steps. The differences sit at acquisition and analysis.
Materials
Disclaimer: This is a general protocol. Clinical laboratories follow institutional SOPs and validated antibody panels (such as EuroFlow [1,2] or BSH-recommended panels [7]). Always check antibody datasheets and optimize for your sample type.
- Whole blood, anticoagulated with EDTA (preferred), sodium heparin, or ACD
- Flow cytometry staining buffer (PBS with BSA or FBS)
- 12 × 75 mm round-bottom polystyrene tubes
- Fluorochrome-labeled antibodies against the markers of interest
- Phosphate-buffered saline (PBS)
- Pipettes, vortex mixer, ice
- Live/dead viability dye (e.g., 7-AAD, propidium iodide, or a fixable amine-reactive dye such as LIVE/DEAD or Zombie series)
- RBC lysis buffer (e.g., ammonium chloride-based)
- Permeabilization buffer (e.g., saponin-based, for intracellular markers)
- Blocking buffer (e.g., FcR block or normal serum)
- Optional: secondary antibodies (for indirect labeling)
- Optional: 40–70 µm cell strainer
- Optional: commercial fix/lyse solution
Protocol
Choosing your workflow: clinical vs research
Before staining, decide which workflow you are running. Clinical labs typically use stain-lyse-no-wash (or stain-lyse-wash) on whole blood. The blood is stained first, the red cells are lysed, and the sample is acquired. This preserves all white cells in their native proportions and minimizes losses [1,7]. Research labs more often isolate peripheral blood mononuclear cells (PBMCs) by density gradient centrifugation (e.g., Ficoll-Paque). PBMC isolation removes granulocytes and red cells but is more cell-friendly for downstream functional assays.
Pick the workflow that matches your goal. The remaining steps are similar.
Sample preparation
Good sample preparation does three things: it isolates the cells of interest, it removes debris that would generate background, and it preserves marker expression.
- For PBMC isolation, layer blood over Ficoll-Paque, centrifuge per manufacturer's instructions, and collect the buffy coat at the interface. For whole-blood workflows, skip directly to lysis after staining.
- Wash twice with 2 mL of staining buffer at 250 × g for 5 minutes. Aspirate carefully so you do not disturb the pellet.
- Count cells and adjust to your target concentration (commonly 1 × 10⁶ cells per 100 µL).
- Handle gently. Avoid bubbles, vigorous vortexing, and over-spinning.
Live/dead staining
Dead cells bind antibodies non-specifically, generate false positives, and clog gates. Live/dead staining lets you exclude them.
- Add a viability dye per manufacturer's instructions. Common choices: 7-AAD or PI (non-fixable); LIVE/DEAD or Zombie series (fixable, amine-reactive).
- Incubate 15 minutes at room temperature or 4°C, in the dark.
- Wash twice with 2 mL of staining buffer at 250 × g for 5 minutes.
Function of Annexin V
Annexin V detects apoptosis, not viability. It marks early apoptotic cells by binding externalized phosphatidylserine. Annexin V is not interchangeable with the viability dyes above.
Surface antibody staining
This is the core step for most clinical immunophenotyping.
For direct labeling (the routine clinical approach):
- Add directly conjugated antibodies at the recommended concentration.
- Incubate 30 minutes at 4°C in the dark.
- Wash twice with staining buffer.
For indirect labeling (less common; used when no direct conjugate exists):
- Add unconjugated primary antibody; incubate 30 minutes.
- Wash twice.
- Add fluorochrome-conjugated secondary antibody; incubate 30 minutes in the dark.
- Wash twice.
When building multicolor panels, choose fluorochromes whose emission spectra do not heavily overlap, and use single-color compensation controls and FMO (fluorescence-minus-one) controls to set accurate gates.
Fixation and permeabilization (for intracellular markers only)
Skip this section if you are staining surface markers only.
Cell membranes block antibodies from reaching intracellular antigens. To stain inside, the membrane must be made porous and the cell must be stabilized so it does not fall apart.
- Fixation crosslinks proteins and stabilizes the cell. Common fixatives: paraformaldehyde (most general), methanol (good for nuclear and phospho-proteins), and glutaraldehyde (cytoskeletal).
- Permeabilization opens the membrane. Saponin and digitonin are gentle and reversible; Triton X-100 is harsher. Methanol and acetone fix and permeabilize in one step.
Order of Process
Standard order when both surface and intracellular markers are needed: surface stain → fix → permeabilize → intracellular stain → wash → acquire.
| Fixative | Time | Temperature | Suitable For | Permeabilization |
|---|---|---|---|---|
| Paraformaldehyde 4% | 10–20 min | 4 °C | Most cytoplasmic and nuclear proteins | Saponin or digitonin |
| Formaldehyde 4% | 10–20 min | 4 °C | General | Triton X-100 or NP-40 |
| Methanol 100% | 5–10 min | −20 °C | Nuclear, phospho-proteins | Not required |
| Acetone 100% | 5–10 min | −20 °C | Cytoplasmic, membrane | Not required |
| Glutaraldehyde 0.25% | 15–30 min | 4 °C | Cytoskeletal | Triton X-100 |
Fixation can mask some epitopes; always confirm with antibody datasheets and run pilot experiments.
Compensation, controls, and acquisition
Compensation corrects for the fact that fluorochromes emit across overlapping wavelength ranges. In conventional flow cytometry, the cytometer subtracts the spillover from each detector based on single-color controls (one tube per fluorochrome, plus an unstained tube). In spectral flow cytometry, the same idea is implemented as spectral unmixing, where the full emission spectrum of each fluorochrome is computed and separated.
Required controls for any clinical or research panel:
- Unstained cells to define autofluorescence.
- Single-color compensation controls (or reference spectra in spectral systems).
- FMO controls to set the boundary between positive and negative for dim markers.
- Isotype controls when nonspecific binding is a concern.
Acquire data at a flow rate that keeps event rate stable. For MRD, collect at least 500,000 to 1,000,000 events per tube to reach the required sensitivity [3,4].
Interpretation

CD45 vs side scatter gating
This plot is the gold standard for analyzing peripheral blood and bone marrow. CD45 is the leukocyte common antigen, expressed at different intensities by different white cells; side scatter reflects granularity.
- Lymphocytes: high CD45, very low SSC.
- Monocytes: high CD45, intermediate SSC.
- Granulocytes: intermediate-to-low CD45, very high SSC.
- Blasts: dim CD45, low SSC. This "blast window" is essential for catching acute leukemia.
- Erythroid precursors: variable CD45, high SSC.
- Plasma cells: low to intermediate CD45, variable SSC.
- Hematopoietic progenitor cells (HPCs): low CD45, low SSC.
Sequential gating
To pull out a specific subset such as T-helper cells:
- Singlet gate (FSC-H vs FSC-A) to exclude doublets.
- Viability gate to exclude dead cells.
- CD45 vs SSC gate to select lymphocytes.
- CD3+ gate to select T cells.
- CD4+ gate for T-helper cells.
Standardized clinical panels
Modern clinical flow cytometry leans heavily on validated panels. The EuroFlow consortium publishes and updates panels for most diagnostic scenarios [1,2,5]. The British Society for Haematology has issued aligned guidelines [7]. Using standardized panels improves reproducibility across labs and sites. To maximize this reproducibility, many clinical laboratories have transitioned to using commercially prepared, lyophilized (freeze-dried) antibody reagent tubes (such as EuroFlow LyoTubes). These pre-mixed, dried formats eliminate liquid pipetting errors, reduce sample preparation time, and ensure absolute consistency between different diagnostic centers [12].
Core Standardized Panels Commonly Utilized in Clinical Laboratories
| Clinical Indication | Key Markers | Target Cells | Diagnostic Use |
|---|---|---|---|
| Acute leukemia orientation (ALOT) | Immaturity: CD34, CD45 (dim), HLA-DR Myeloid: CD117, CD13, CD33, cMPO B-lymphoid: CD19, cCD79a, cCD22 T-lymphoid: cCD3, CD7 | Blasts | Distinguishes AML from B-ALL or T-ALL; identifies mixed-phenotype acute leukemia |
| B-cell chronic lymphoproliferative disorders | Pan-B: CD19, CD20 Subtyping: CD5, CD10, CD23, CD200, FMC7, CD103 Clonality: surface kappa (sκ) and lambda (sλ) | Mature B cells | Subtypes CLL, mantle cell lymphoma, follicular lymphoma |
| T- and NK-cell neoplasms | Pan-T: CD2, CD3, CD4, CD5, CD7, CD8 NK/other: CD56, CD16, CD57, TCR-αβ, TCR-γδ | Mature T and NK cells | Detects loss of pan-T markers, skewed CD4:CD8 ratios, T-LGL leukemia, Sézary syndrome |
| Plasma cell dyscrasias | Identification: CD38, CD138, CD45 (often dim/negative) Aberrancy: CD56, CD117, CD19, CD27, CD81 Clonality: cytoplasmic kappa (cκ) and lambda (cλ) | Plasma cells | Detects monoclonal plasma cells in multiple myeloma, smoldering myeloma, MGUS |
| PNH screening | WBC: CD15, CD24, CD14, CD59, FLAER RBC: CD59, CD235a | Neutrophils, monocytes, RBCs | Confirms loss of GPI-anchored proteins; quantifies PNH clone size [9] |
| MDS assessment | Myeloid/monocytic: CD11b, CD13, CD14, CD16, CD33, CD64, HLA-DR Erythroid: CD71, CD105 | Maturing granulocytes, monocytes, erythroid precursors | Evaluates dysplastic maturation patterns and aberrant antigen expression |
Measurable Residual Disease (MRD)
After induction chemotherapy, a patient may look in remission by morphology yet still harbor a small number of leukemic cells. This is measurable residual disease (MRD) or formerly called minimal residual disease, but renamed because the disease is not minimal in clinical impact [3]. Using optimized Next-Generation Flow (NGF) protocols that acquire millions of events, flow cytometry routinely detects MRD at sensitivities of 10⁻⁵ (1 in 100,000 cells) and can reach limits of 10⁻⁶ in specific diseases like multiple myeloma and B-ALL. This exceptional sensitivity makes modern flow cytometry highly competitive with molecular techniques like Next-Generation Sequencing (NGS), and MRD status is now one of the strongest predictors of relapse in AML and ALL [3,4,8,13].
Two complementary approaches dominate:
- Leukemia-Associated Immunophenotype (LAIP): an aberrant marker combination defined on the patient's own leukemic cells at diagnosis, then tracked at follow-up.
- Difference from Normal (DfN): any deviation from normal hematopoietic maturation patterns, which catches clones that have shifted their phenotype during therapy.
Most modern labs combine both [3,4]. Sample requirements are demanding: fresh bone marrow, at least 500,000 to 1,000,000 acquired events, and analyst expertise.
Applications of Flow Cytometry in Hematology

Flow cytometry's footprint in hematology is wide:
- Diagnosis and classification of leukemias and lymphomas. Different blood cancers carry distinct marker patterns. Flow cytometry is fast, sensitive, and the routine front-line test [1,7].
- MRD monitoring. As above [3,4].
- PNH screening. FLAER plus CD24/CD15 on neutrophils and CD14/CD15 on monocytes confirm GPI-anchored protein deficiency. PNH clones as small as ~1% are clinically meaningful in aplastic anemia [9].
- Immune deficiency workup. Lymphocyte subset counts (CD3, CD4, CD8, CD19, CD16/56) are standard in HIV monitoring and in suspected primary immunodeficiency.
- Transplant compatibility. Flow crossmatch detects donor-specific antibodies before solid organ or hematopoietic cell transplant.
- Research and drug development. Immune cell profiling, apoptosis assays, cell cycle analysis, and signaling pathway studies all rely on flow cytometry.
Other Immunophenotyping Methods
While flow cytometry is the dominant tool, there are other methods for immunophenotyping:
- Immunohistochemistry (IHC): visualizes proteins in fixed tissue sections; gives spatial context but limited multiparameter capacity.
- Mass cytometry (CyTOF): uses metal-tagged antibodies and a mass spectrometer; allows 40+ parameters but cannot sort cells and is slower.
- Imaging flow cytometry: combines flow cytometry with microscopy; gives both quantitative and morphological data per cell.
- Single-cell RNA sequencing: profiles transcriptomes rather than proteins; complements flow cytometry in research.
Frequently Asked Questions (FAQs)
How long can a blood sample be stored before flow cytometry analysis?
Whole blood for routine immunophenotyping should be processed within 24–48 hours of collection, kept at room temperature, and never frozen before processing. Surface markers and viability degrade over time; some markers like CD62L are lost within hours.
What is fluorescence compensation, and why is it needed?
Fluorochromes emit across overlapping wavelength ranges, so light from one dye spills into detectors meant for another. Compensation subtracts that overlap mathematically, using single-color control tubes. In spectral flow cytometry, the equivalent step is called spectral unmixing.
What is the difference between conventional and spectral flow cytometry?
Conventional cytometers detect one peak per fluorochrome through filters and typically support 8–12 colors. Spectral cytometers capture each cell's full emission spectrum and unmix it computationally, supporting 25–40+ colors in one tube [6]. Spectral systems handle small or precious samples particularly well.
What is measurable residual disease, and why has the name changed?
MRD is the small population of leukemia cells remaining after treatment, below the threshold of microscopic remission. The field renamed "minimal" to "measurable" because the residual disease is not minimal in impact as it strongly predicts relapse [3]. Flow cytometry, qPCR, and next-generation sequencing are the main detection methods.
Why is CD45 vs side scatter the standard gating strategy in hematology?
CD45 is expressed at different intensities on different leukocytes. Plotting it against side scatter cleanly separates lymphocytes, monocytes, granulocytes, and blasts. The "dim CD45, low side scatter" blast window is essential for catching acute leukemia.
Can flow cytometry detect all blood disorders?
No. It is excellent for cell-based disorders (leukemias, lymphomas, plasma cell dyscrasias, PNH, immunodeficiencies) but cannot, on its own, detect coagulation disorders, hemoglobinopathies (these need HPLC or genetic testing), or most red cell membrane disorders. Diagnosis usually combines flow cytometry with molecular testing, cytogenetics, and morphology.
Glossary of Related Medical Terms
- Antigen. Any molecule recognized by the immune system. In flow cytometry, usually a surface or intracellular protein bound by a labeled antibody.
- CD (Cluster of Differentiation). A standardized naming system for cell-surface markers (e.g., CD3 for T cells, CD45 for leukocytes).
- Compensation. A correction that removes signal spilling from one fluorochrome into another's detector.
- Difference from Normal (DfN). An MRD approach that looks for any deviation from normal maturation patterns.
- EuroFlow. A European consortium that publishes validated, standardized 8-color panels for diagnosing blood cancers [1,2].
- Fluorochrome. A fluorescent dye conjugated to an antibody (e.g., FITC, PE, APC, BV421).
- FMO control. A sample stained with every antibody in a panel except one, used to set the boundary between positive and negative for that one marker.
- Forward Scatter (FSC). Light scattered forward by a cell; tracks cell size.
- Gating. Drawing boundaries on a plot to select a population for further analysis.
- LAIP. Leukemia-Associated Immunophenotype is an aberrant marker combination defined at diagnosis and tracked for MRD.
- Measurable Residual Disease (MRD). Very small numbers of leukemia cells remaining after treatment, detectable by flow cytometry, qPCR, or NGS [3].
- Side Scatter (SSC). Light scattered sideways; tracks internal complexity.
- Spectral flow cytometry. A flow cytometry approach that captures the full emission spectrum of each cell and unmixes it computationally [6].
- Spillover spread. Residual noise added to a detector by a spillover fluorochrome, even after compensation. Limits detection of dim markers.
Disclaimer: This protocol is for educational purposes only. Local laboratory standard operating procedures take precedence. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. Always consult a qualified healthcare professional for clinical decision-making. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Kalina, T., Flores-Montero, J., van der Velden, V. H., Martin-Ayuso, M., Böttcher, S., Ritgen, M., Almeida, J., Lhermitte, L., Asnafi, V., Mendonça, A., de Tute, R., Cullen, M., Sedek, L., Vidriales, M. B., Pérez, J. J., te Marvelde, J. G., Mejstrikova, E., Hrusak, O., Szczepański, T., van Dongen, J. J., … EuroFlow Consortium (EU-FP6, LSHB-CT-2006-018708) (2012). EuroFlow standardization of flow cytometer instrument settings and immunophenotyping protocols. Leukemia, 26(9), 1986–2010. https://doi.org/10.1038/leu.2012.122
- van Dongen, J. J., Lhermitte, L., Böttcher, S., Almeida, J., van der Velden, V. H., Flores-Montero, J., Rawstron, A., Asnafi, V., Lécrevisse, Q., Lucio, P., Mejstrikova, E., Szczepański, T., Kalina, T., de Tute, R., Brüggemann, M., Sedek, L., Cullen, M., Langerak, A. W., Mendonça, A., Macintyre, E., … EuroFlow Consortium (EU-FP6, LSHB-CT-2006-018708) (2012). EuroFlow antibody panels for standardized n-dimensional flow cytometric immunophenotyping of normal, reactive and malignant leukocytes. Leukemia, 26(9), 1908–1975. https://doi.org/10.1038/leu.2012.120
- Heuser, M., Freeman, S. D., Ossenkoppele, G. J., Buccisano, F., Hourigan, C. S., Ngai, L. L., Tettero, J. M., Bachas, C., Baer, C., Béné, M. C., Bücklein, V., Czyz, A., Denys, B., Dillon, R., Feuring-Buske, M., Guzman, M. L., Haferlach, T., Han, L., Herzig, J. K., Jorgensen, J. L., … Cloos, J. (2021). 2021 Update on MRD in acute myeloid leukemia: a consensus document from the European LeukemiaNet MRD Working Party. Blood, 138(26), 2753–2767. https://doi.org/10.1182/blood.2021013626
- Tettero, J. M., Freeman, S., Buecklein, V., Venditti, A., Maurillo, L., Kern, W., Walter, R. B., Wood, B. L., Roumier, C., Philippé, J., Denys, B., Jorgensen, J. L., Bene, M. C., Lacombe, F., Plesa, A., Guzman, M. L., Wierzbowska, A., Czyz, A., Ngai, L. L., Schwarzer, A., … Buccisano, F. (2021). Technical Aspects of Flow Cytometry-based Measurable Residual Disease Quantification in Acute Myeloid Leukemia: Experience of the European LeukemiaNet MRD Working Party. HemaSphere, 6(1), e676. https://doi.org/10.1097/HS9.0000000000000676
- Holl, E., Kapinsky, M., & Larbi, A. (2025). An Update on Flow Cytometry Analysis of Hematological Malignancies: Focus on Standardization. Cancers, 17(12), 2045. https://doi.org/10.3390/cancers17122045
- Sharma, S., Boyer, J., & Teyton, L. (2024). A practitioner's view of spectral flow cytometry. Nature methods, 21(5), 740–743. https://doi.org/10.1038/s41592-023-02042-3
- Johansson, U., Bloxham, D., Couzens, S., Jesson, J., Morilla, R., Erber, W., Macey, M., & British Committee for Standards in Haematology (2014). Guidelines on the use of multicolour flow cytometry in the diagnosis of haematological neoplasms. British Committee for Standards in Haematology. British journal of haematology, 165(4), 455–488. https://doi.org/10.1111/bjh.12789
- Döhner, H., Wei, A. H., Appelbaum, F. R., Craddock, C., DiNardo, C. D., Dombret, H., Ebert, B. L., Fenaux, P., Godley, L. A., Hasserjian, R. P., Larson, R. A., Levine, R. L., Miyazaki, Y., Niederwieser, D., Ossenkoppele, G., Röllig, C., Sierra, J., Stein, E. M., Tallman, M. S., Tien, H. F., … Löwenberg, B. (2022). Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood, 140(12), 1345–1377. https://doi.org/10.1182/blood.2022016867
- Borowitz, M. J., Craig, F. E., Digiuseppe, J. A., Illingworth, A. J., Rosse, W., Sutherland, D. R., Wittwer, C. T., Richards, S. J., & Clinical Cytometry Society (2010). Guidelines for the diagnosis and monitoring of paroxysmal nocturnal hemoglobinuria and related disorders by flow cytometry. Cytometry. Part B, Clinical cytometry, 78(4), 211–230. https://doi.org/10.1002/cyto.b.20525
- Saeys, Y., Van Gassen, S., & Lambrecht, B. N. (2016). Computational flow cytometry: helping to make sense of high-dimensional immunology data. Nature reviews. Immunology, 16(7), 449–462. https://doi.org/10.1038/nri.2016.56
- Park, L. M., Lannigan, J., & Jaimes, M. C. (2020). OMIP-069: Forty-Color Full Spectrum Flow Cytometry Panel for Deep Immunophenotyping of Major Cell Subsets in Human Peripheral Blood. Cytometry. Part A : the journal of the International Society for Analytical Cytology, 97(10), 1044–1051. https://doi.org/10.1002/cyto.a.24213
- van Dongen, J. J., Orfao, A., & EuroFlow Consortium (2012). EuroFlow: Resetting leukemia and lymphoma immunophenotyping. Basis for companion diagnostics and personalized medicine. Leukemia, 26(9), 1899–1907. https://doi.org/10.1038/leu.2012.121
- Flores-Montero, J., Sanoja-Flores, L., Paiva, B., Puig, N., García-Sánchez, O., Böttcher, S., van der Velden, V. H. J., Pérez-Morán, J. J., Vidriales, M. B., García-Sanz, R., Jimenez, C., González, M., Martínez-López, J., Corral-Mateos, A., Grigore, G. E., Fluxá, R., Pontes, R., Caetano, J., Sedek, L., Del Cañizo, M. C., … Orfao, A. (2017). Next Generation Flow for highly sensitive and standardized detection of minimal residual disease in multiple myeloma. Leukemia, 31(10), 2094–2103. https://doi.org/10.1038/leu.2017.29