Key Takeaways
A platelet (thrombocyte) is a small, disc-shaped cell fragment, 2–4 µm wide, made in the bone marrow from megakaryocytes; the normal count is 150,000–450,000/µL and each platelet lives 7–10 days.
- Primary Function (Hemostasis): Platelets stop bleeding through three steps: adhesion to the injured vessel wall via von Willebrand factor, activation with shape change and release of granules (ADP, thromboxane A2, serotonin, calcium), and aggregation through fibrinogen bridges between GPIIb/IIIa receptors [1].
- Key Regulation: Thrombopoietin (TPO), made mainly in the liver, is the master regulator of platelet production; healthy endothelium balances clotting by releasing nitric oxide and prostacyclin to keep platelets quiet.
- Thrombocytosis (High) ▾: Thrombocytosis (high platelets) is usually reactive, but primary forms like essential thrombocythemia carry a real risk of thrombosis.
- Thrombocytopenia (Low) ▾: Thrombocytopenia (low platelets) presents with mucosal bleeding, petechiae, and easy bruising; common causes include immune thrombocytopenia (ITP), heparin-induced thrombocytopenia (HIT), thrombotic thrombocytopenic purpura (TTP), bone marrow failure, and splenic sequestration [2,3,5].
- Key Labs ▾: Core investigations are the complete blood count (with platelet count and MPV), peripheral blood smear, and platelet function tests such as light transmission aggregometry; bleeding time is largely outdated.
*Click ▾ for more information
Introduction
Platelets are the smallest cells in your blood, but they do some of the heaviest lifting. When a blood vessel breaks, platelets are the first responders, plugging the gap within seconds before the slower clotting proteins finish the job. They also act as messengers in inflammation, infection, and wound healing [8].
This article walks you through what platelets look like, how they work, how the body makes them, and what happens when their numbers or function go wrong.
What Platelets Look Like and How They Are Built
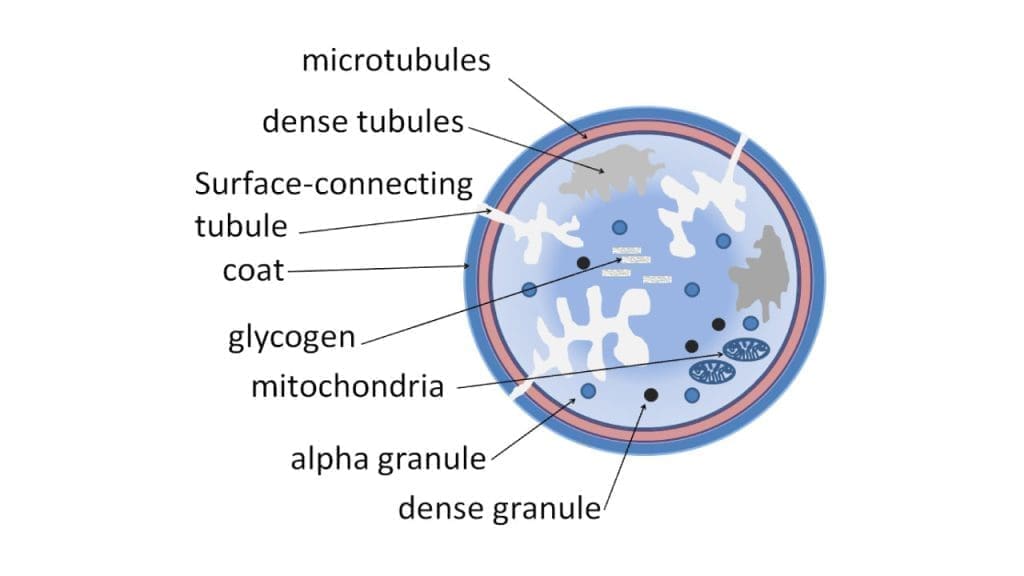
A resting platelet is a smooth disc, 2–4 µm across, with no nucleus. The disc shape is not decoration; it lets platelets glide through small vessels without sticking to healthy walls and gives them a large surface area relative to their volume, ready to deploy on contact with injury.

The platelet's outer coat, called the glycocalyx, carries a negative charge that repels healthy endothelial cells. When the vessel wall is damaged and collagen is exposed, this same surface flips into "attach" mode through specialized receptors:
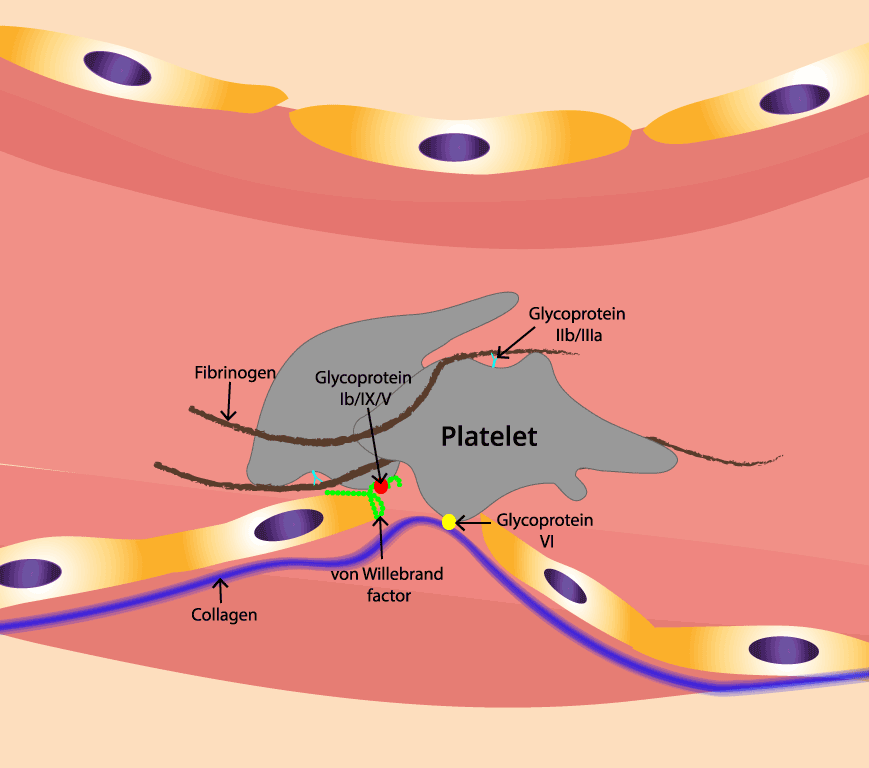
- GPIb-IX-V binds von Willebrand factor (vWF), a long protein that bridges the platelet to exposed collagen. This is the very first contact.
- Integrins like GPIIb/IIIa (αIIbβ3) become the strong, long-lasting anchors. They bind fibrinogen, the bridge that links one platelet to another.
Inside the platelet sit two important storage compartments. Alpha granules, the more numerous of the two (roughly 50–80 per platelet), contain fibrinogen, vWF, factor V, and growth factors. Dense granules (about 3–8 per platelet) are smaller but punchier, packed with ADP, ATP, calcium, and serotonin. When a platelet activates, it dumps these granules into the surrounding plasma, calling more platelets to the site.
Platelets also carry a few mitochondria for energy and a network of microtubules and actin filaments (the cytoskeleton) that lets them shape-shift from disc to spiky sphere within seconds of activation [1].
How Platelets Stop Bleeding: Adhesion, Activation, Aggregation
Primary hemostasis happens in three quick steps. Each one matters, and each one can fail in a different disease.
Step 1: Adhesion
When the endothelium breaks, collagen underneath is exposed to flowing blood. vWF in plasma binds to that collagen and uncoils under shear stress, exposing sticky regions that grab the platelet's GPIb-IX-V receptor. This first grip is fast but weak — a "tether and roll" — which gives the platelet time to sense the surface and switch on its stronger anchors [9].
Step 2: Activation
Once a platelet attaches, it gets multiple "go" signals: collagen, ADP from damaged cells, thromboxane A2 from neighboring platelets, and thrombin from the coagulation cascade. Each signal works through a receptor that triggers a calcium spike inside the platelet. Calcium drives:
- Shape change — disc to spiky sphere, with finger-like pseudopods that grip nearby surfaces.
- Granule release — alpha and dense granules empty their cargo, recruiting more platelets.
- Receptor activation — GPIIb/IIIa flips from a folded "off" shape into an extended "on" shape that can bind fibrinogen.
Aspirin works at this stage by permanently blocking cyclooxygenase-1, the enzyme platelets use to make thromboxane A2. Because platelets cannot synthesize new enzymes, a single dose of aspirin dampens the platelet for its entire lifespan [1].
Step 3: Aggregation
Activated GPIIb/IIIa receptors on one platelet bind fibrinogen, and the other end of that fibrinogen molecule binds GPIIb/IIIa on a neighboring platelet. As more platelets activate and join, fibrinogen bridges accumulate and the loose pile becomes a tight plug. The coagulation cascade then converts fibrinogen into fibrin, which weaves a tough mesh around the plug and seals the wound.
Built-In Brakes
Healthy endothelium does not let this run wild. It releases two natural brakes: nitric oxide (NO) and prostacyclin (PGI2). Both raise levels of cyclic nucleotides inside platelets, which suppress activation, calcium signaling, and shape change. The result is a clear rule of the road — platelets stay calm next to healthy vessels and only switch on at the site of injury.
Thrombopoiesis
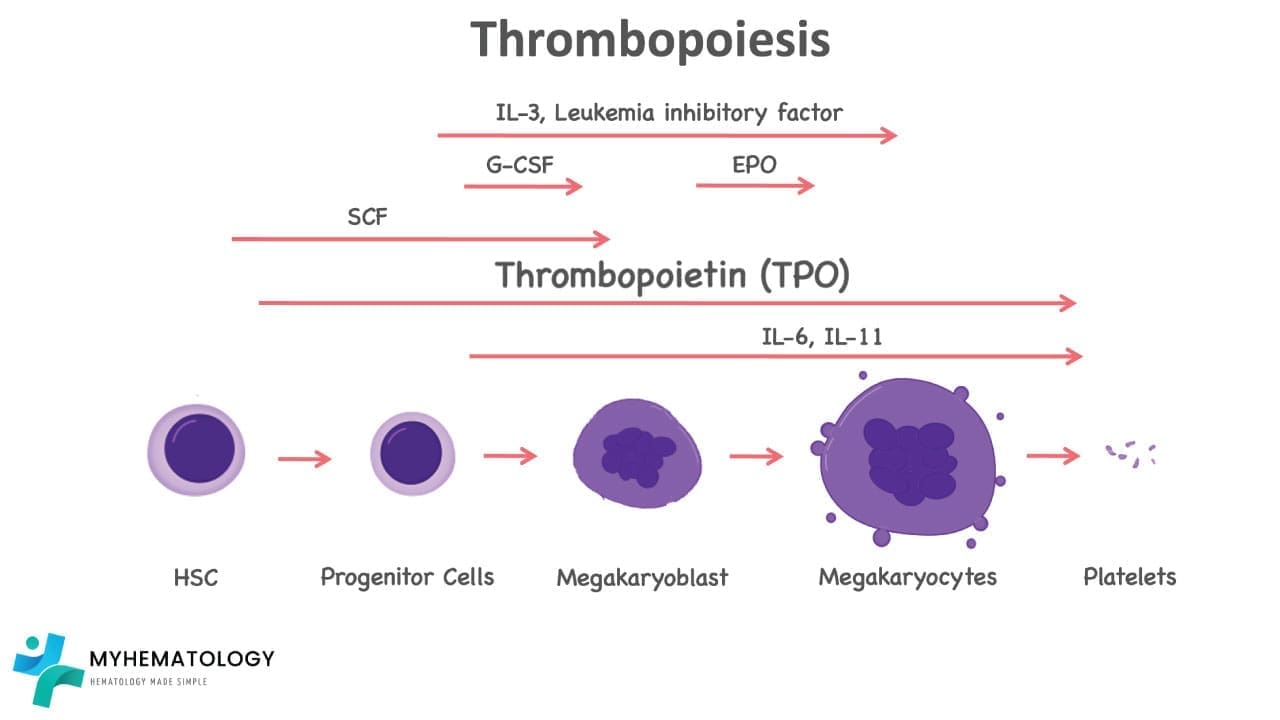
Platelets are made in the bone marrow from giant cells called megakaryocytes. The process unfolds in stages:
- A hematopoietic stem cell commits to the megakaryocyte lineage.
- The cell grows enormous and undergoes endomitosis (repeated DNA replication without cell division) producing a single multilobed nucleus with up to 64 sets of chromosomes.
- The cytoplasm fills with proplatelet ribbons, long extensions that reach into bone marrow blood vessels.
- Shear forces in the sinusoids snap these ribbons into individual platelets that enter circulation.
A single megakaryocyte can release roughly 1,000–3,000 platelets before it dies.
The hormone thrombopoietin (TPO) runs the show. TPO is made mainly by the liver, with smaller contributions from the kidney and bone marrow. Aged platelets carry desialylated glycoproteins that signal the liver to make more TPO. This is an elegant feedback loop that keeps the count steady [10]. Other cytokines like IL-3, IL-6, and stem cell factor support megakaryocyte growth, especially during stress.
This is why patients with cirrhosis often have low platelets: less TPO from the liver, plus an enlarged spleen that traps them.
Platelets Beyond Clotting
For decades, hematology textbooks framed platelets purely as clot-makers. That picture is incomplete. Platelets carry over 1,000 different proteins in their granules and can sense pathogens directly through Toll-like receptors. They release signals that recruit neutrophils, support neutrophil extracellular trap (NET) formation, and regulate macrophage behavior at sites of infection [8].
Practical implications:
- Platelets play a role in sepsis, where unchecked thromboinflammation can damage organs.
- They influence wound healing through growth factors like PDGF and TGF-β — the basis of platelet-rich plasma therapy.
- They contribute to atherosclerosis and tumor metastasis by interacting with the vessel wall and circulating tumor cells.
High Platelet Count: Thrombocytosis
A platelet count above 450,000/µL is called thrombocytosis. The clinical question is always: reactive or primary?
Reactive (secondary) thrombocytosis is far more common. Triggers include:
- Inflammation (rheumatoid arthritis, inflammatory bowel disease)
- Infection
- Iron deficiency anemia
- Recovery from surgery, trauma, or major bleeding
- Post-splenectomy state
- Malignancy
Reactive thrombocytosis usually resolves once the trigger is treated and rarely causes thrombosis on its own.
Primary thrombocytosis comes from a problem inside the bone marrow. The leading example is essential thrombocythemia (ET), a myeloproliferative disorder driven in many cases by mutations in JAK2, CALR, or MPL. ET carries a real risk of arterial and venous thrombosis, and sometimes paradoxical bleeding when counts climb very high. Familial thrombocytosis is a rare genetic cause.
Most people with thrombocytosis have no symptoms; the count is often discovered on a routine blood test.
Low Platelet Count: Thrombocytopenia
A platelet count below 150,000/µL is thrombocytopenia. The cause falls into one of three buckets, and identifying which bucket matters more than the count itself.
1. Decreased Production
The bone marrow is not making enough platelets. Causes include aplastic anemia, leukemia or lymphoma infiltrating the marrow, chemotherapy and radiation, viral infections (HIV, hepatitis C), severe vitamin B12 or folate deficiency, and inherited disorders.
2. Increased Destruction
Platelets are being made normally but destroyed too quickly. Examples:
- Immune thrombocytopenia (ITP) — the immune system makes antibodies against the body's own platelets [2,3].
- Heparin-induced thrombocytopenia (HIT) — antibodies form against complexes of heparin and platelet factor 4, causing both low platelets and paradoxical thrombosis [5,6].
- Thrombotic thrombocytopenic purpura (TTP) — a deficiency of the enzyme ADAMTS13 leads to ultra-large vWF multimers that chew through platelets, causing microvascular clots, hemolysis, and end-organ damage. This is a hematologic emergency [11].
- Disseminated intravascular coagulation (DIC) — widespread clotting consumes platelets and clotting factors.
- Vaccine-induced immune thrombotic thrombocytopenia (VITT) — a rare reaction initially identified with adenoviral vector COVID-19 vaccines. While largely historical now due to shifts in vaccine platforms, the study of VITT fundamentally reshaped hematology by revealing how anti-PF4 antibodies can spontaneously cause thrombosis, leading to the recognition of similar non-vaccine anti-PF4 syndromes [12,13].
3. Sequestration
Platelets are pooled (trapped) in an enlarged spleen. Common in cirrhosis, portal hypertension, and infiltrative spleen diseases.
Pseudo-Thrombocytopenia
Sometimes the platelet count looks low on the analyzer but is actually normal. The classic cause is EDTA-dependent platelet clumping, where the anticoagulant in the purple-top tube triggers antibody-mediated clumping. The fix is to repeat the count in a citrated (blue-top) tube. Other causes include platelet satellitism (platelets sticking to neutrophils), giant platelets in inherited disorders like Bernard-Soulier syndrome, and partial clotting in a poorly drawn sample. Always look at the smear before making major treatment decisions.
Spotting Platelet Disorders Clinically
Platelet bleeding looks different from coagulation factor bleeding, and learning the difference is one of the most useful clinical pattern recognition skills.
Platelet-type bleeding (low or dysfunctional platelets):
- Petechiae and purpura on the skin
- Mucosal bleeding — gums, nose, heavy menstrual bleeding
- Bleeding immediately after a cut or procedure
- Gastrointestinal bleeding in severe cases
Coagulation factor bleeding (e.g., hemophilia):
- Deep bleeding into joints (hemarthrosis) and muscles
- Delayed bleeding after trauma
- Large hematomas
Thrombotic complications of platelet disorders include deep vein thrombosis (DVT), pulmonary embolism, stroke, and myocardial infarction — relevant especially in essential thrombocythemia, HIT, TTP, and VITT.
Laboratory Investigations for Platelet Evaluation
A focused workup uses a small set of tests:
- Complete Blood Count (CBC) — gives the platelet count (normal 150–450 × 10⁹/L), mean platelet volume or MPV (about 7.5–10.5 fL), and platelet distribution width.
- Peripheral blood smear — confirms the count, looks for clumping, giant platelets, schistocytes (suggesting TTP/DIC), and abnormal white cells. Always check the smear before treating thrombocytopenia.
- Light transmission aggregometry (LTA) — the gold standard for platelet function, testing how platelets clump in response to agonists like ADP, collagen, epinephrine, and ristocetin.
- PFA-100/200 (Platelet Function Analyzer) — a faster screen that has largely replaced the older bleeding time test.
- Immature platelet fraction (IPF) — analogous to a reticulocyte count for platelets; high IPF suggests increased production (destruction or consumption), low IPF suggests marrow failure.
- Coagulation panel (PT, aPTT, INR) — to separate platelet problems from clotting factor problems.
- Specific tests when indicated — anti-platelet antibodies (limited utility in ITP), HIT antibody assays plus serotonin release assay, ADAMTS13 activity for TTP, JAK2/CALR/MPL mutations for myeloproliferative neoplasms, and bone marrow biopsy when production is in question.
Management of Platelet Disorders
Management depends entirely on the cause. The same low platelet count can be observed in one patient and trigger an emergency response in another.
Immune Thrombocytopenia (ITP)
The 2019 ASH guidelines, reviewed in 2022, recommend [3,4]:
- First-line: short courses of corticosteroids (prednisone or dexamethasone), preferred over longer courses to reduce side effects. IV immunoglobulin (IVIG) for rapid response when bleeding is significant.
- Second-line: TPO receptor agonists e.g. eltrombopag, romiplostim, or avatrombopag, are now preferred over splenectomy or rituximab for many adults. Avatrombopag, FDA-approved in 2019, is taken orally without food restrictions, unlike eltrombopag.
- Emerging therapies: FcRn inhibitors (such as efgartigimod) have recently entered the ITP landscape. These drugs work by accelerating the degradation of pathogenic IgG autoantibodies, offering a novel, targeted mechanism for refractory cases [14].
- Splenectomy is generally deferred at least 12–24 months from diagnosis to allow for spontaneous remission, and is reserved for patients who fail medical therapy.
Heparin-Induced Thrombocytopenia (HIT)
HIT is a "platelet-low, clot-high" disorder. The diagnostic approach uses the 4Ts score [5,6,7]:
A score of 0–3 has a negative predictive value near 99.8% and effectively rules HIT out [7]. Scores of 4–5 (intermediate) and ≥6 (high) require antibody testing and a confirmatory functional assay (serotonin release or PF4-induced flow cytometry).
If HIT is suspected, stop all heparin immediately including flushes and start a non-heparin anticoagulant. Direct oral anticoagulants (DOACs), particularly rivaroxaban and apixaban, are now widely accepted as frontline therapies for hemodynamically stable patients due to their efficacy and ease of use compared to intravenous options like argatroban or bivalirudin [5,6,15]. Do not give platelet transfusions unless the patient is bleeding; they may worsen thrombosis.
Thrombotic Thrombocytopenic Purpura (TTP)
TTP is a hematologic emergency. The classic pentad — thrombocytopenia, microangiopathic hemolytic anemia, fever, neurological symptoms, and renal dysfunction — is rarely all present. Modern care includes [11]:
- Therapeutic plasma exchange (started urgently, often before confirmatory ADAMTS13 results)
- Corticosteroids
- Caplacizumab, an anti-vWF nanobody, FDA-approved in 2019, which shortens time to platelet recovery and reduces TTP-related death and recurrence
- Rituximab for refractory or relapsing immune-mediated disease
- Recombinant ADAMTS13 (apadamtase alfa) was FDA-approved in late 2023 for congenital TTP (cTTP), this provides targeted enzyme replacement therapy and marks a major milestone. Acquired immune TTP still relies primarily on plasma exchange and caplacizumab [16].
Platelet Transfusion Thresholds
General guidance for prophylactic platelet transfusion:
- ≤10 × 10⁹/L — most stable hospitalized patients with thrombocytopenia
- ≤20 × 10⁹/L — fever, sepsis, or planned minor procedures
- ≤50 × 10⁹/L — most surgical procedures, lumbar puncture
- ≤100 × 10⁹/L — neurosurgery or ocular surgery
Transfusions are usually avoided in HIT, TTP, and ITP unless bleeding is life-threatening, because they can fuel the underlying disease.
Supportive Care
Patients with low platelets benefit from practical measures: avoiding contact sports and high-fall-risk activities, soft toothbrushes and electric razors, no NSAIDs or aspirin without medical advice, and prompt reporting of new bruising, petechiae, or unusual headaches. For caregivers, knowing what counts as an emergency — sudden severe headache, neurological change, uncontrolled bleeding, blood in stool or urine — is often more useful than the number on the lab report.
A Brief Historical Note
The story of the platelet started with what 18th-century anatomists called "milky particles." In 1882, the Italian physician Giulio Bizzozero observed live platelets in frog blood and named them piastrine, "little plates." The American pathologist James Homer Wright coined "thrombocyte" in 1906 after showing their role in clotting. The 20th-century arrival of electron microscopy, the discovery of vWF, and the mapping of platelet receptors transformed a curiosity into one of the best-characterized cells in the body — and opened the door to today's antiplatelet drugs and TPO-based therapies [10].
Frequently Asked Questions (FAQs)
What is a platelet and what does it do?
A platelet is a small cell fragment in your blood, about 2–4 micrometers wide, made in the bone marrow from large parent cells called megakaryocytes. Platelets stop bleeding by sticking to injured blood vessels, clumping with each other, and releasing chemicals that strengthen the clot. They also help with wound healing and immune defense.
What is a normal platelet count, and what counts are dangerous?
A normal platelet count is 150,000–450,000 per microliter (150–450 × 10⁹/L). Counts below 50,000/µL raise concern for surgical bleeding. Spontaneous bleeding usually does not occur until counts fall below about 20,000/µL, and counts under 10,000/µL are considered critically low. Above 450,000/µL is called thrombocytosis, and very high counts can raise clotting risk.
What is the difference between thrombocytopenia and thrombocytosis?
Thrombocytopenia means a low platelet count (below 150,000/µL) and tends to cause bleeding, bruising, petechiae, or nosebleeds. Thrombocytosis means a high platelet count (above 450,000/µL) and can either be a reaction to inflammation, infection, or iron deficiency, or be primary, as in essential thrombocythemia. Primary thrombocytosis carries a real risk of thrombosis.
Why does taking aspirin affect platelets?
Aspirin permanently blocks an enzyme called cyclooxygenase-1 inside platelets, which they need to make thromboxane A2. Without thromboxane A2, platelets cannot fully activate or recruit other platelets. Because platelets cannot make new enzymes, this effect lasts for the platelet's entire 7–10 day lifespan, which is why aspirin is taken once daily for cardiovascular protection.
What is heparin-induced thrombocytopenia (HIT)?
HIT is a serious immune reaction in which the body makes antibodies against complexes of heparin and platelet factor 4. Despite the platelet count falling, these antibodies activate platelets and cause clots, not bleeding. It typically appears 5–10 days after starting heparin. Clinicians use the 4Ts score to estimate likelihood, confirm with antibody testing, stop all heparin, and switch to a non-heparin anticoagulant such as argatroban or fondaparinux.
Can a person live a normal life with a platelet disorder?
Yes, in many cases. Many people with mild thrombocytopenia or controlled thrombocytosis live full, active lives with monitoring and lifestyle adjustments such as avoiding contact sports, careful medication choices, and good dental care. Severe disorders like ITP, TTP, or inherited platelet disorders need specialist care, but modern treatments including TPO receptor agonists, immunotherapy, and targeted drugs like caplacizumab for TTP have transformed outcomes.
Glossary of Related Medical Terms
- Adhesion — the first step of clotting, where platelets stick to the damaged blood vessel wall using von Willebrand factor as a bridge.
- Aggregation — platelets sticking to each other, forming a plug. Held together mainly by fibrinogen.
- Alpha granules — the larger and more numerous storage compartments in a platelet, containing fibrinogen, von Willebrand factor, and growth factors.
- Dense granules — smaller storage compartments containing ADP, ATP, calcium, and serotonin, all of which amplify platelet activation.
- Endothelium — the single-cell lining of blood vessels. Healthy endothelium prevents platelets from sticking; damaged endothelium activates them.
- Fibrinogen — a plasma protein that forms bridges between activated platelets and is later converted to fibrin, the meshwork that stabilizes a clot.
- GPIb-IX-V — the receptor on platelets that grabs onto von Willebrand factor and starts the adhesion process.
- GPIIb/IIIa (αIIbβ3) — the receptor that binds fibrinogen and links platelets to one another during aggregation.
- Hemostasis — the body's process for stopping bleeding.
- Megakaryocyte — the large bone marrow cell that breaks apart to release platelets.
- Mean platelet volume (MPV) — the average size of platelets, reported in femtoliters (fL).
- Petechiae — pinpoint red or purple spots on the skin caused by small bleeds; classic sign of low platelets.
- Platelet (thrombocyte) — a tiny, anucleate cell fragment that helps stop bleeding and supports immune and healing functions.
- Thrombocytopenia — low platelet count, below 150,000/µL.
- Thrombocytosis — high platelet count, above 450,000/µL.
- Thrombopoietin (TPO) — the hormone made mainly by the liver that drives platelet production.
- Thrombosis — unwanted clot formation inside a blood vessel.
- Thromboxane A2 (TXA2) — a lipid signal released by activated platelets that recruits more platelets and narrows blood vessels.
- Von Willebrand factor (vWF) — a plasma and endothelial protein that anchors platelets to injured vessel walls.
Disclaimer: This article is intended for educational and informational purposes only. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Neunert, C. E., & Cooper, N. (2018). Evidence-based management of immune thrombocytopenia: ASH guideline update. Hematology. American Society of Hematology. Education Program, 2018(1), 568–575. https://doi.org/10.1182/asheducation-2018.1.568
- Provan, D., Arnold, D. M., Bussel, J. B., Chong, B. H., Cooper, N., Gernsheimer, T., Ghanima, W., Godeau, B., González-López, T. J., Grainger, J., Hou, M., Kruse, C., McDonald, V., Michel, M., Newland, A. C., Pavord, S., Rodeghiero, F., Scully, M., Tomiyama, Y., Wong, R. S., … Kuter, D. J. (2019). Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood advances, 3(22), 3780–3817. https://doi.org/10.1182/bloodadvances.2019000812
- Neunert, C., Terrell, D. R., Arnold, D. M., Buchanan, G., Cines, D. B., Cooper, N., Cuker, A., Despotovic, J. M., George, J. N., Grace, R. F., Kühne, T., Kuter, D. J., Lim, W., McCrae, K. R., Pruitt, B., Shimanek, H., & Vesely, S. K. (2019). American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood advances, 3(23), 3829–3866. https://doi.org/10.1182/bloodadvances.2019000966
- Neunert, C. E., Arnold, D. M., Grace, R. F., Kuhne, T., McCrae, K. R., & Terrell, D. R. (2024). The 2022 review of the 2019 American Society of Hematology guidelines on immune thrombocytopenia. Blood advances, 8(13), 3578–3582. https://doi.org/10.1182/bloodadvances.2023012541
- Cuker, A., Arepally, G. M., Chong, B. H., Cines, D. B., Greinacher, A., Gruel, Y., Linkins, L. A., Rodner, S. B., Selleng, S., Warkentin, T. E., Wex, A., Mustafa, R. A., Morgan, R. L., & Santesso, N. (2018). American Society of Hematology 2018 guidelines for management of venous thromboembolism: heparin-induced thrombocytopenia. Blood advances, 2(22), 3360–3392. https://doi.org/10.1182/bloodadvances.2018024489
- May, J., & Cuker, A. (2024). Practical guide to the diagnosis and management of heparin-induced thrombocytopenia. Hematology. American Society of Hematology. Education Program, 2024(1), 388–395. https://doi.org/10.1182/hematology.2024000566
- Cuker, A., Gimotty, P. A., Crowther, M. A., & Warkentin, T. E. (2012). Predictive value of the 4Ts scoring system for heparin-induced thrombocytopenia: a systematic review and meta-analysis. Blood, 120(20), 4160–4167. https://doi.org/10.1182/blood-2012-07-443051
- Mandel, J., Casari, M., Stepanyan, M., Martyanov, A., & Deppermann, C. (2022). Beyond Hemostasis: Platelet Innate Immune Interactions and Thromboinflammation. International journal of molecular sciences, 23(7), 3868. https://doi.org/10.3390/ijms23073868
- Scharf R. E. (2018). Platelet Signaling in Primary Haemostasis and Arterial Thrombus Formation: Part 1. Hamostaseologie, 38(4), 203–210. https://doi.org/10.1055/s-0038-1675144
- Kaushansky K. (2008). Historical review: megakaryopoiesis and thrombopoiesis. Blood, 111(3), 981–986. https://doi.org/10.1182/blood-2007-05-088500
- Scully, M., Cataland, S. R., Peyvandi, F., Coppo, P., Knöbl, P., Kremer Hovinga, J. A., Metjian, A., de la Rubia, J., Pavenski, K., Callewaert, F., Biswas, D., De Winter, H., Zeldin, R. K., & HERCULES Investigators (2019). Caplacizumab Treatment for Acquired Thrombotic Thrombocytopenic Purpura. The New England journal of medicine, 380(4), 335–346. https://doi.org/10.1056/NEJMoa1806311
- Greinacher, A., Thiele, T., Warkentin, T. E., Weisser, K., Kyrle, P. A., & Eichinger, S. (2021). Thrombotic Thrombocytopenia after ChAdOx1 nCov-19 Vaccination. The New England journal of medicine, 384(22), 2092–2101. https://doi.org/10.1056/NEJMoa2104840
- Warkentin, T. E., & Greinacher, A. (2023). Laboratory Testing for Heparin-Induced Thrombocytopenia and Vaccine-Induced Immune Thrombotic Thrombocytopenia Antibodies: A Narrative Review. Seminars in thrombosis and hemostasis, 49(6), 621–633. https://doi.org/10.1055/s-0042-1758818
- Broome, C., Miyakawa, Y., Carpenedo, M., Al-Samkari, H., Ayguasanosa, J., Phillips, J., & Rodeghiero, F. (2026). Efgartigimod as a treatment for adults with primary immune thrombocytopenia: a plain language summary of the ADVANCE IV study. Therapeutic advances in hematology, 17, 20406207261445668. https://doi.org/10.1177/20406207261445668
- Davis, K. A., & Davis, D. O. (2017). Direct acting oral anticoagulants for the treatment of suspected heparin-induced thrombocytopenia. European journal of haematology, 99(4), 332–335. https://doi.org/10.1111/ejh.12921
- Scully, M., Knöbl, P., Kentouche, K., Rice, L., Windyga, J., Schneppenheim, R., Kremer Hovinga, J. A., Kajiwara, M., Fujimura, Y., Maggiore, C., Doralt, J., Hibbard, C., Martell, L., & Ewenstein, B. (2017). Recombinant ADAMTS-13: first-in-human pharmacokinetics and safety in congenital thrombotic thrombocytopenic purpura. Blood, 130(19), 2055–2063. https://doi.org/10.1182/blood-2017-06-788026