TL;DR
Autoimmune Hemolytic Anemia (AIHA) is a condition where the body’s immune system mistakenly attacks and destroys its own red blood cells, leading to anemia. This destruction process is known as hemolysis.
- Types ▾:
- Warm AIHA
- Cold Agglutinin Disease (CAD)
- Paroxysmal Cold Hemoglobinuria (PCH)
- Mixed AIHA
- Drug-induced AIHA
- Symptoms ▾:
- Fatigue
- Weakness
- Shortness of breath
- Pale skin
- Jaundice
- Dark urine
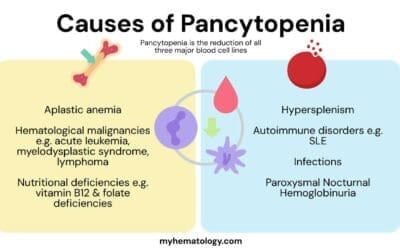
- Complications ▾:
- Severe anemia
- Increased risk of infections
- Blood clots
- Heart failure
- Diagnosis ▾:
- Complete blood count (CBC) – anemia
- Peripheral blood smear – spherocytes (warm AIHA), agglutination (CAD)
- Direct Antiglobulin Test (DAT) – positive
- Cold agglutination test (for CAD) – positive
- Bilirubin – elevated
- Lactate dehydrogenase (LDH) – elevated
- Differential Diagnosis ▾:
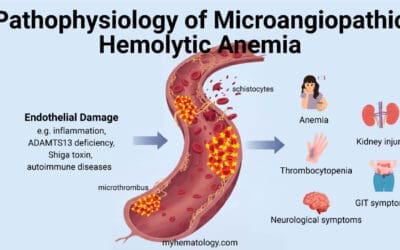
- Microangiopathic hemolytic anemia (MAHA)
- Paroxysmal nocturnal hemoglobinuria (PNH)
- Hereditary hemolytic anemias
- Vitamin deficiencies
- Drug-induced hemolysis
- Sepsis
- Underlying malignancies
- Treatment ▾
- Corticosteroids are first-line therapy
- Rituximab,
- Supportive care for anemia
- Addressing underlying causes
*Click ▾ for more information
What is autoimmune hemolytic anemia?
Autoimmune hemolytic anemia (AIHA) is a rare blood disorder where the body’s immune system mistakenly attacks its own red blood cells.
Red blood cells are essential for carrying oxygen throughout the body. In autoimmune hemolytic anemia (AIHA), the immune system produces antibodies that target these red blood cells, causing them to break down prematurely leading to hemolysis and hemolytic anemia.
What is hemolysis?
Hemolysis refers to the breakdown of red blood cells (RBCs), leading to the release of their internal contents, primarily hemoglobin, into the surrounding plasma.
Hemolysis can occur within the bloodstream (intravascular hemolysis) or after RBCs are taken up by macrophages in the spleen and liver (extravascular hemolysis).
Clinical Significance of Hemolysis
- Anemia: The primary consequence of hemolysis is a decrease in the number of functional RBCs, leading to anemia. This translates to symptoms like fatigue, weakness, and shortness of breath due to impaired oxygen delivery to tissues.
- Jaundice: Free hemoglobin released during hemolysis is broken down by the liver into bilirubin, a yellow pigment. Excessive bilirubin accumulation can cause jaundice, a yellowing of the skin and sclerae (whites of the eyes).
- Hemoglobinuria: In severe hemolysis, the kidneys may not be able to handle the high amount of free hemoglobin, leading to its presence in the urine (hemoglobinuria), giving it a dark reddish-brown color.
Intravascular vs Extravascular Hemolysis
| Feature | Intravascular Hemolysis | Extravascular Hemolysis |
| Site of destruction | Within bloodstream | Outside bloodstream (primarily spleen) |
| Cause | Direct attack on RBC membrane by complement or immune cells | Phagocytosis of RBCs coated with antibodies by macrophages in the spleen |
| Initiating factors | Severe infections, toxins, incompatible blood transfusions | Autoimmune diseases, hereditary RBC defects |
| Hemoglobin release | Rapid release of free hemoglobin and iron into plasma | Gradual release of hemoglobin following RBC breakdown by macrophages |
| Clinical presentation | More severe symptoms due to rapid hemolysis (hemoglobinuria, shock) | Less severe symptoms initially, may progress to jaundice and anemia |
| Examples | Severe transfusion reactions, hemolytic uremic syndrome | Autoimmune hemolytic anemia (AIHA), hereditary spherocytosis |
Types of AIHA
- Warm AIHA: This is the most common type (around 70% of cases). In warm autoimmune hemolytic anemia (AIHA), the antibodies attacking red blood cells are active at body temperature (37°C or higher). These antibodies are typically IgG and sometimes IgA. They bind to red blood cells, marking them for destruction by macrophages in the spleen (extravascular hemolysis).
- Cold agglutinin disease (CAD): Less common than warm autoimmune hemolytic anemia (AIHA) (around 30% of cases). Here, the antibodies are active at cooler temperatures (below 37°C). These antibodies are primarily IgM and can cause red blood cells to clump together (agglutinate) at cooler extremities like fingers and toes. This agglutination can block blood flow and lead to tissue damage.
- Paroxysmal cold hemoglobinuria (PCH): PCH is a rare type of autoimmune hemolytic anemia (AIHA) characterized by a sudden onset of symptoms triggered by exposure to cold temperatures.
- Mixed AIHA: A rare type where the patient has both warm and cold antibodies active, causing features of both warm and cold autoimmune hemolytic anemia (AIHA).
- Drug-Induced AIHA (DIIHA): While a cause, the mechanism of hemolysis often falls into warm or cold characteristics depending on the specific drug and antibody.
| Type of AIHA | Antibody Type | Optimal Activity Temp. | Primary Mechanism of Hemolysis | Direct Antiglobulin Test (DAT) | Key Clinical Features |
| Warm AIHA (WAIHA) | IgG | (body temp) | Extravascular (spleen) | Positive for IgG ( ± C3d) | Acute/subacute anemia, fatigue, pallor, jaundice, splenomegaly |
| Cold Agglutinin Disease (CAD) | IgM | 0−4∘C (cold) | Intravascular (complement) | Positive for C3d ( ± IgM) | Chronic anemia, acrocyanosis, Raynaud’s phenomenon, hemoglobinuria (cold exposure) |
| Mixed-Type AIHA | IgG and IgM | Both warm and cold | Both extravascular & intravascular | Positive for IgG & C3d | Severe anemia, features of both WAIHA and CAD |
| Paroxysmal Cold Hemoglobinuria (PCH) | Biphasic IgG (Donath-Landsteiner) | Binds cold, lyses warm | Intravascular (complement) | Positive for C3d only | Acute, severe hemolytic crisis post-cold exposure, fever, chills, dark urine |
| Drug-Induced AIHA | IgG, IgM (variable) | Variable (often warm) | Variable (hapten, immune complex, autoantibody induction) | Variable (IgG, C3d, or both) | Resembles WAIHA or intravascular hemolysis; resolves with drug discontinuation |
Causes of AIHA
- Primary AIHA (Idiopathic): In about half of autoimmune hemolytic anemia (AIHA) cases, no underlying cause can be identified. This is called primary or idiopathic AIHA.
- Secondary AIHA: In the other half of cases, autoimmune hemolytic anemia (AIHA) develops alongside another underlying condition that triggers the immune system to attack red blood cells. Some common causes of secondary AIHA include:
- Infections (e.g., Epstein-Barr virus, cytomegalovirus)
- Autoimmune diseases (e.g., systemic lupus erythematosus, Sjögren’s syndrome)
- Certain medications
- Cancers (lymphoma, leukemia)
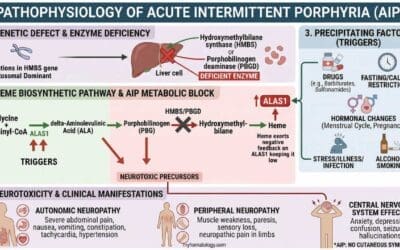
Pathophysiology of AIHA
The pathophysiology of Autoimmune Hemolytic Anemia (AIHA) revolves around a fundamental breakdown in immune tolerance, leading the body’s own immune system to mistakenly attack and destroy its red blood cells (RBCs). This complex process involves specific immune components and distinct mechanisms of RBC destruction.
Immune System Dysfunction
At the core of AIHA is a failure of the immune system to distinguish between “self” and “non-self” antigens. Normally, the immune system is highly regulated to prevent attacks on the body’s own tissues. In AIHA, this regulation goes awry.
Loss of Self-Tolerance
This can occur through various mechanisms, including genetic predispositions, environmental triggers (like infections or drugs), or dysregulation of T-regulatory cells that normally suppress autoimmune responses.
Role of Complement System

The complement system is a collection of over 20 plasma proteins that work together to enhance the immune system’s ability to clear pathogens and damaged cells. It acts as a powerful but tightly regulated cascade system, meaning its effects amplify as the pathway progresses.
Functions of the Complement System
- Opsonization: Complement proteins coat the surface of pathogens or damaged cells (like antibody-bound red blood cells in AIHA), making them more recognizable and easier for phagocytes (white blood cells like macrophages) to engulf and destroy.
- Inflammatory Response: Complement activation fragments can trigger inflammation, attracting immune cells to the site of infection or tissue damage.
- Direct Lysis: In some cases, the complement system can directly create pores in the membrane of pathogens, leading to their lysis (bursting). This is particularly important for eliminating certain bacteria.
Activation Pathways
The complement system can be activated through several pathways, each triggered by different molecules.
- Classical Pathway: Activated by antigen-antibody complexes (e.g., IgG bound to a pathogen). This is the most common pathway involved in autoimmune hemolytic anemia (AIHA) (warm type).
- Lectin Pathway: Triggered by binding of mannan-binding lectin (MBL) to specific sugar groups on pathogens or apoptotic (dying) cells. IgM antibodies in cold autoimmune hemolytic anemia (AIHA) can also activate this pathway.
- Alternative Pathway: Can be activated by various surfaces, including microbial membranes, without the need for antibodies. It plays a role in providing a backup mechanism for complement activation.
Complement Cascade

Once a pathway is initiated, a cascade of enzymatic reactions occurs. Complement proteins cleave and activate other complement proteins in a sequential manner, amplifying the immune response.
Key Complement Components
- C1q: (Classical pathway) Recognizes and binds to the Fc region of antibodies attached to pathogens.
- C3b and C4b: These opsonizing fragments are deposited on the target surface, marking it for phagocytosis.
- Membrane Attack Complex (MAC): Formed by a series of late-stage complement proteins, it can create pores in the membrane of target cells, leading to lysis.
Regulation of the Complement System
The complement system is tightly regulated to prevent damage to healthy host tissues. Several control proteins act at various points in the cascade to prevent inappropriate activation and limit potential tissue injury.
Regulation at Multiple Levels
Complement regulation occurs throughout the activation cascade, ensuring control at various points to minimize the risk of unintended consequences.
Plasma Regulatory Proteins
Several regulatory proteins present in the blood plasma act as “brakes” on the complement system:
- Membrane Inhibitory Proteins (MIPs): These proteins bind to C3b and C4b, preventing their further activation and subsequent formation of the Membrane Attack Complex (MAC).
- Complement Factor H (FH): This protein cleaves C3b, inactivating its ability to bind to target surfaces and initiate the assembly of MAC.
- Complement Factor I (FI): Works in conjunction with FH, further cleaving C3b to enhance its inactivation.
- Decay-Accelerating Factors (DAFs): These proteins accelerate the dissociation of C3 convertase enzymes (C4b2a and C3bBb), preventing them from generating excessive C3b and halting the cascade.
Cell-Surface Regulatory Proteins
- Membrane Cofactor Protein (MCP): Present on host cells, MCP acts as a cofactor for FH, facilitating C3b inactivation on cell surfaces and preventing self-targeting by the complement system.
- CD59: This protein directly inhibits the formation of MAC on host cell membranes, protecting them from lysis.
Regulation by Complement Receptors
- CR1 (Complement Receptor 1): Found on various immune cells like erythrocytes and macrophages, CR1 binds to C3b on target surfaces but doesn’t initiate a strong inflammatory response. This allows for controlled phagocytosis of opsonized cells without excessive complement activation.
Consequences of Dysregulation
Disruptions in these regulatory mechanisms can lead to uncontrolled complement activation, potentially contributing to various diseases, including:
- Autoimmune diseases: Unintended complement activation can target healthy tissues, as seen in autoimmune hemolytic anemia (AIHA) where antibodies mark red blood cells for destruction by the complement system.
- Inflammatory disorders: Excessive complement activation can lead to uncontrolled inflammation and tissue damage.
Warm Autoimmune Hemolytic Anemia (AIHA)
The key characteristic of warm AIHA is that the autoantibodies involved are active at body temperature (around 37°C or higher). This temperature dependence differentiates warm autoimmune hemolytic anemia (AIHA) from cold agglutinin disease.
Why Body Temperature Matters?
The temperature at which autoantibodies bind to RBCs determines the mechanism of destruction.
- Warm AIHA: Antibodies are active at body temperature. This allows them to bind to RBCs throughout the bloodstream.
- Cold Agglutinin Disease (CAD): Antibodies are active at cooler temperatures. Binding primarily occurs in extremities where blood flow is slower and temperatures are lower.
Mechanism of Warm AIHA
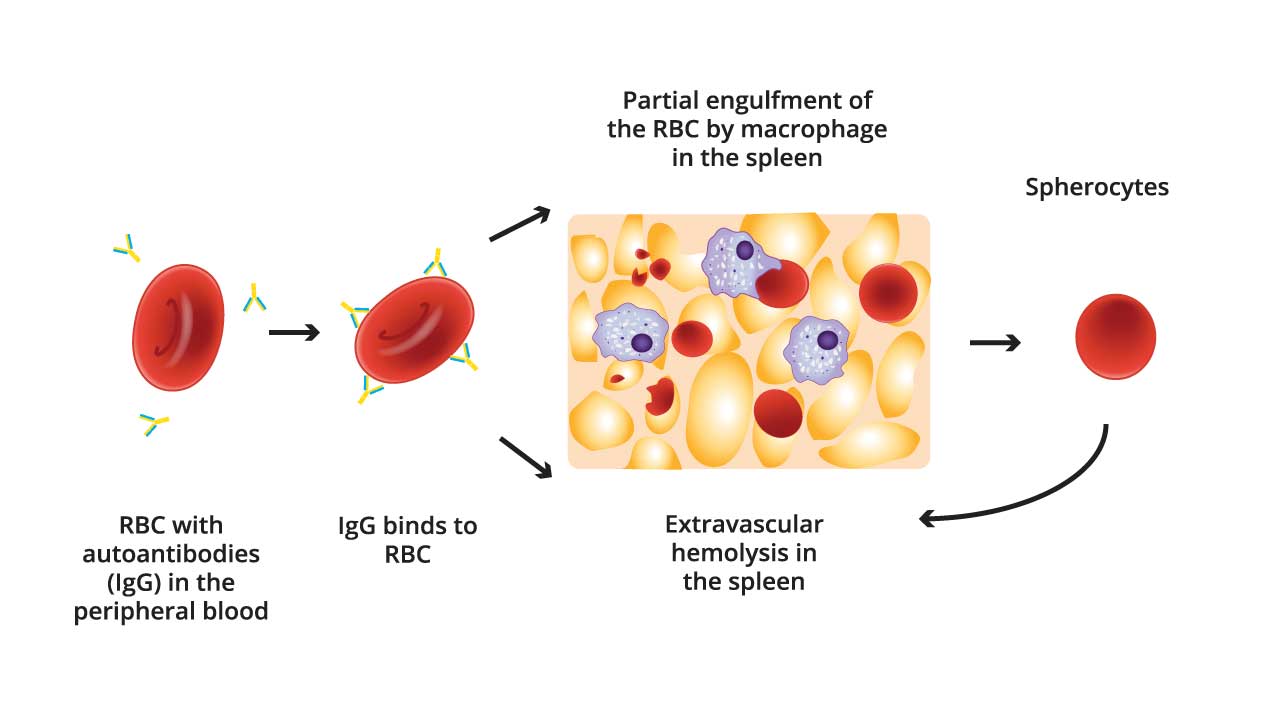
- Autoantibody Production: B lymphocytes produce IgG antibodies (sometimes IgA) that target antigens on healthy RBCs.
- Antibody Binding: These IgG autoantibodies bind to the red blood cell antigens with high affinity. This creates an “opsonic” effect, essentially tagging the red blood cells for destruction by immune cells.
- Fc Receptor Recognition: The Fc region of the bound IgG antibody can be recognized by Fc receptors present on macrophages.
- Complement Activation (Classical Pathway) (not always necessary): The Fc region of bound IgG antibodies can activate the classical pathway of the complement system. C1q, the first component, recognizes and binds to the Fc region.
- Opsonization: Enzymatic reactions in the complement cascade lead to the deposition of C3b and C4b fragments onto the RBC surface. These fragments mark the RBCs for destruction by phagocytes.
- Phagocytosis by Macrophages: Macrophages have Fc receptors specifically designed to recognize the Fc region of antibodies. Upon binding to the Fc portion of the IgG on the red blood cell surface, the macrophage engulfs and destroys the red blood cell through a process called phagocytosis. Macrophages also have receptors for C3b and C4b, which recognize opsonized RBCs and engulf them.
- Hemolysis: The engulfed RBCs are broken down inside the macrophages, releasing hemoglobin and cellular components into the circulation. This breakdown is hemolysis.
Additional Points
- The type and amount of autoantibodies produced can influence the severity of warm autoimmune hemolytic anemia (AIHA).
- The spleen is the primary site of RBC destruction due to the high concentration of macrophages present there.
- Not all IgG-coated RBCs are immediately destroyed. The lifespan of these cells depends on the specific antibody and complement interactions.
Cold Agglutinin Disease (CAD)
Cold agglutinin disease (CAD) is another form of autoimmune hemolytic anemia, but with a key distinction – the autoantibodies involved are active at cooler temperatures (below 37°C). This temperature dependence has significant implications for the mechanism of RBC destruction in cold agglutinin disease (CAD) compared to warm autoimmune hemolytic anemia (AIHA).
Importance of Body Temperature
Warm autoimmune hemolytic anemia (AIHA) antibodies can bind to RBCs throughout the body because they’re active at body temperature. In cold autoimmune hemolytic anemia (AIHA), the cooler temperature requirement for antibody binding leads to a different scenario.
- Binding primarily occurs in extremities: Blood flow is slower and temperatures are lower in hands, feet, and ears. This is why symptoms like cold-induced acrocyanosis (bluish discoloration of extremities) can be seen in cold agglutinin disease.
- Agglutination (clumping) of RBCs: The cold antibodies cause RBCs to clump together, particularly in cooler areas. These clumps can block blood flow and lead to tissue damage in the affected areas.
Mechanism of Cold Agglutinin Disease (CAD)
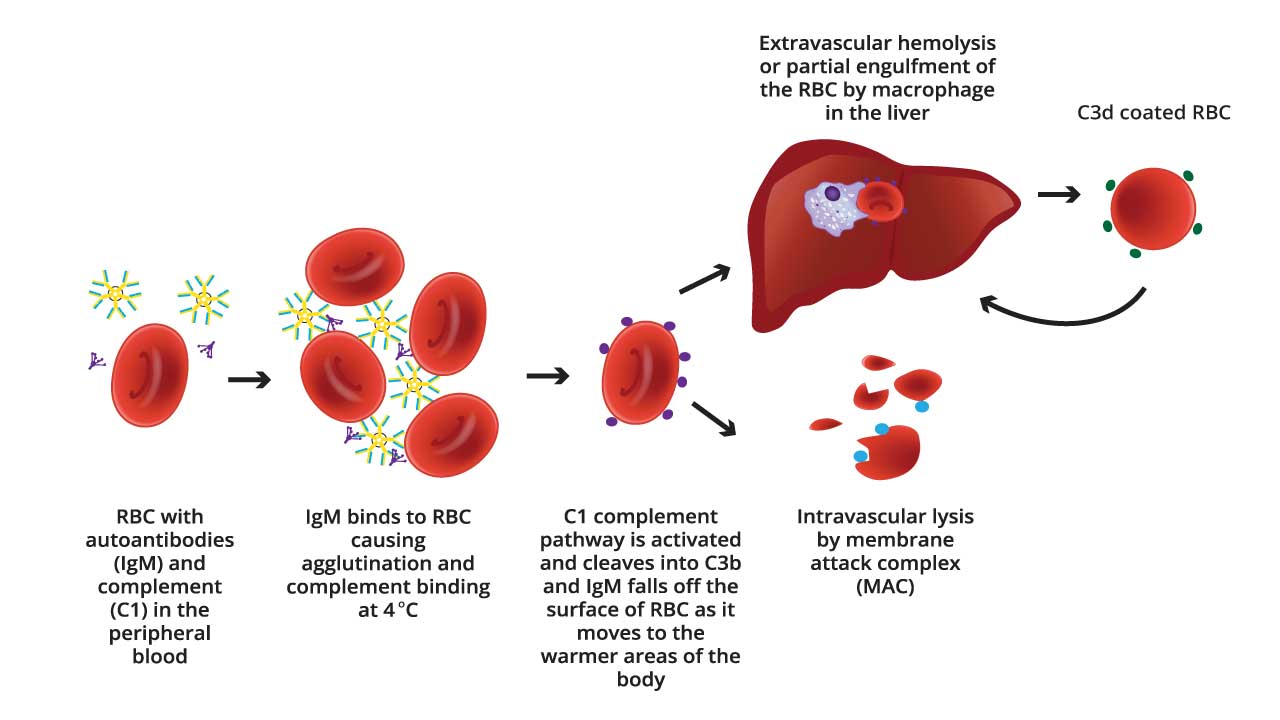
IgM in CAD activates the complement system through a less efficient interaction with C1q compared to IgG in warm AIHA. This often leads to sublytic complement activation and opsonization, promoting phagocytosis by macrophages as the primary mechanism of red blood cell destruction.
IgM and the Classical Complement Pathway
- Antibody Binding: Cold agglutinins (IgM autoantibodies) bind to specific I/i antigens on the surface of red blood cells at cooler temperatures (below body temperature).
- C1q Binding: Unlike IgG, IgM antibodies generally do not directly bind to C1q, the first component of the classical complement pathway. However, the size and structure of the pentameric IgM can sometimes facilitate a low-efficiency interaction with C1q.
- Alternative Complement Pathway Activation (Potential): In some cases, IgM bound to the red blood cell surface might activate the alternative complement pathway through a protein called C3b. However, this is a less efficient and slower process compared to the classical pathway.
- Limited Complement Cascade: Even if C1q binds to the IgM, the subsequent steps in the classical complement pathway activation are often less efficient compared to IgG-mediated activation. This is because the structure of IgM doesn’t optimally facilitate the conformational changes needed for efficient activation of later complement components (C4 and C2).
Sublytic Complement Activation
Despite the limitations, IgM-mediated complement activation in CAD often leads to a sublytic attack on the red blood cell. This means the complement cascade doesn’t progress all the way to form the Membrane Attack Complex (MAC) that directly lyses the cell.
Mechanisms of Red Blood Cell Clearance
- C3b Opsonization: Complement fragments like C3b generated during the limited complement activation can opsonize (tag) the red blood cell surface.
- Enhanced Phagocytosis: Macrophages in the spleen recognize these C3b tags through complement receptors and engulf the IgM-coated red blood cells through phagocytosis.
- Extravascular Hemolysis: Inside the macrophages, the red blood cells are broken down, leading to their destruction, a process called extravascular hemolysis.
Additional Points
- The severity of cold agglutinin disease (CAD) can vary depending on the strength of the cold agglutinins and the extent of RBC agglutination.
- Unlike warm autoimmune hemolytic anemia (AIHA), cold agglutinins may not always directly activate the full complement cascade, but they can still mark RBCs for destruction by macrophages.
- Cold agglutination can sometimes be observed during blood testing in cold AIHA, providing a clue to the diagnosis.
General Signs and Symptoms (common to both warm AIHA and CAD)
Anemia is the core consequence of autoimmune hemolytic anemia (AIHA), as the immune system destroys healthy RBCs, reducing their ability to carry oxygen throughout the body. Symptoms of anemia can include:
- Fatigue and weakness
- Shortness of breath
- Pale skin
- Dizziness or lightheadedness
- Rapid heartbeat (tachycardia)
Additional Signs and Symptoms
- Jaundice: When RBCs are broken down, they release hemoglobin, a yellow pigment. Excessive bilirubin, a breakdown product of hemoglobin, can accumulate in the bloodstream and cause yellowing of the skin and sclerae (whites of the eyes).
- Dark urine: Breakdown products of hemoglobin are excreted in the urine, which can appear dark brown or reddish.
- Spleen enlargement (splenomegaly): The spleen is the primary site for extravascular hemolysis in AIHA. An enlarged spleen might be palpable on physical examination in some cases.
Symptoms Specific to CAD
- Acrocyanosis: Bluish discoloration of the skin due to cold-induced vasoconstriction, particularly in the extremities (hands and feet).
- Livedo reticularis: A lacy red-purple pattern on the skin, often seen on the legs, caused by agglutination of RBCs in small blood vessels.
Complications of Autoimmune Hemolytic Anemia (AIHA)
AIHA (Autoimmune Hemolytic Anemia) can lead to various complications if left untreated.
General Complications
- Severe Anemia: Uncontrolled destruction of red blood cells can lead to significant anemia, causing extreme fatigue, shortness of breath, and heart problems as the body struggles to deliver oxygen to tissues.
- High Blood Output Heart Failure: To compensate for anemia, the heart may work harder to pump more blood, potentially leading to high-output heart failure in severe cases.
- Increased Risk of Infections: Anemia can weaken the immune system, making individuals more susceptible to infections.
- Thrombosis (Blood Clots): Damage to red blood cells and inflammation associated with AIHA can increase the risk of blood clots forming in veins or arteries. These clots can block blood flow and lead to serious complications like stroke, pulmonary embolism (blood clot in the lung), or deep vein thrombosis (DVT). This risk is more pronounced in warm AIHA.
Complications Specific to CAD
Skin Ulceration and Gangrene: Severe cases of CAD, particularly those with very strong cold agglutinins, can lead to tissue damage and even skin ulceration or gangrene in the extremities due to prolonged cold-induced vasoconstriction and impeded blood flow caused by RBC agglutination.
Laboratory Investigations for AIHA
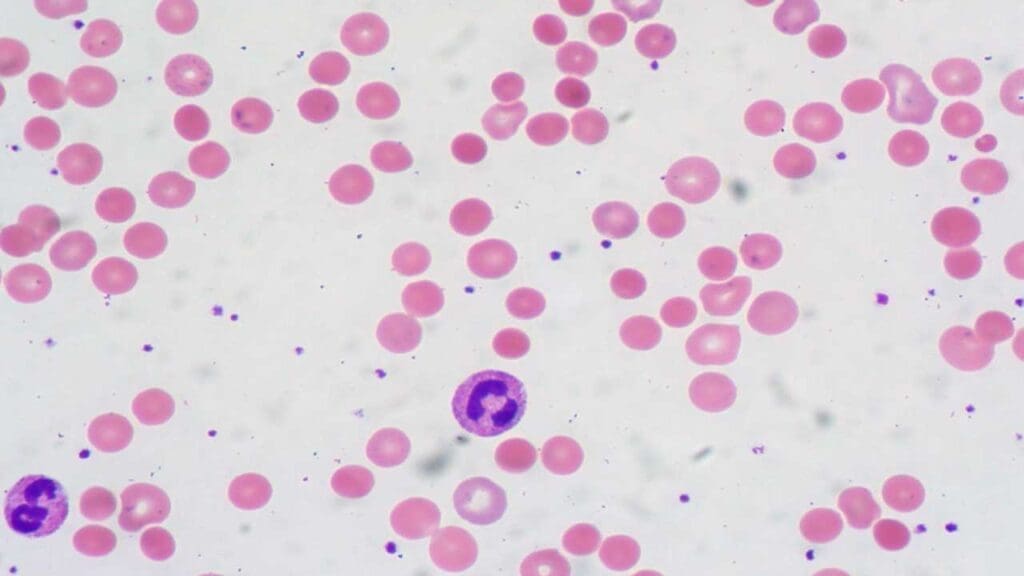
Laboratory investigations play a vital role in diagnosing AIHA (Autoimmune Hemolytic Anemia) and pinpointing the specific type, whether warm or cold. Here’s a comparison of key investigations and their expected results.
| Test | Description | Warm AIHA | Cold Agglutinin Disease (CAD) |
| Complete Blood Count (CBC) | Measures red blood cell (RBC) count, hemoglobin, hematocrit, and other blood cell parameters. | Anemia (low RBC count, hemoglobin, and hematocrit) May show increased reticulocytes (immature RBCs) indicating the bone marrow is trying to compensate for RBC destruction. | Anemia (low RBC count, hemoglobin, and hematocrit) Reticulocyte count may be normal or slightly elevated. |
| Peripheral Blood Smear | Examines the size, shape, and overall health of RBCs under a microscope. | May show spherocytes (round, sphere-shaped RBCs) due to macrophage destruction in the spleen. | May show agglutination (clumping) of RBCs, especially at the edges of the smear. |
| Direct Antiglobulin Test (DAT or Coombs’ test) | Detects the presence of antibodies or complement proteins bound to RBCs. Positive test is a hallmark of AIHA. | DAT positive for IgG, sometimes IgA, indicating antibodies coating the RBCs. | Complement DAT positive for IgM, indicating cold agglutinins bound to RBCs. |
| Cold Agglutination Test | Specifically assesses for the presence and temperature dependence of cold agglutinins. | Usually negative or weakly positive. | Positive agglutination at cooler temperatures (below body temperature), confirming cold agglutinins. Titer (dilution at which agglutination occurs) can indicate severity. |
| Other Tests | May include tests to identify underlying conditions associated with AIHA (e.g., viral serology, autoimmune disease workup). | Elevated serum bilirubin Elevated serum lactate dehydrogenase (LDH) | Elevated serum bilirubin Elevated serum lactate dehydrogenase (LDH) |
Differential Diagnosis for AIHA
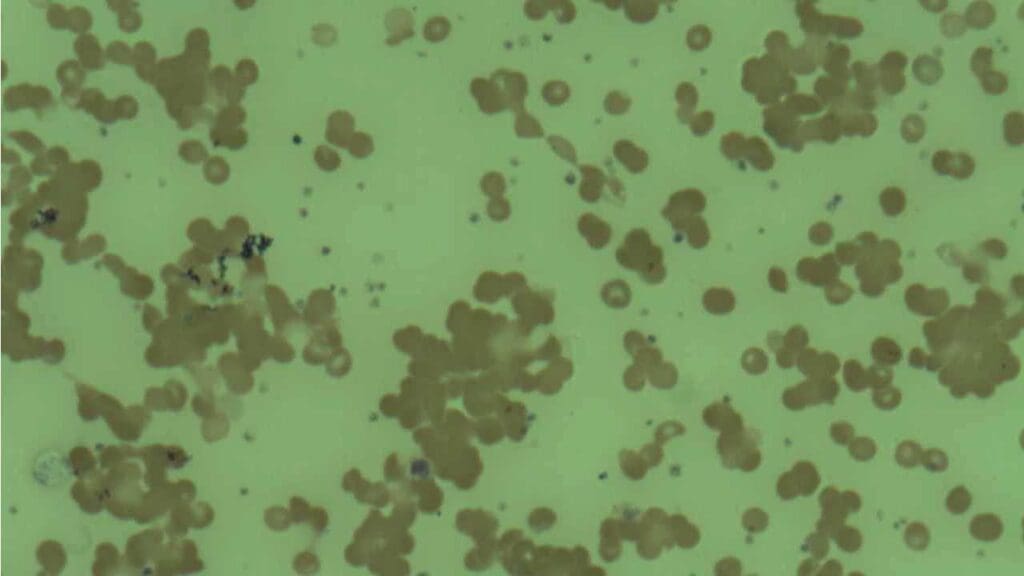
AIHA (Autoimmune Hemolytic Anemia) can present with symptoms similar to other conditions that cause red blood cell destruction.
A thorough medical history, physical examination, laboratory investigations like CBC, blood smear, DAT, and potentially other tests depending on the suspected differential diagnosis, are essential to differentiate AIHA from other causes of hemolytic anemia.
| Differential Diagnosis | Key Differentiating Features | Relevant Diagnostic Tests |
| Alloimmune Hemolysis | History of recent transfusion (transfusion reaction) or pregnancy (HDFN). Often acute onset. | Positive DAT (often mixed field or poly-specific, depending on cause), positive IAT, crossmatch incompatibility for transfusion reactions. |
| Microangiopathic Hemolytic Anemias (MAHA) | Presence of schistocytes (fragmented red blood cells) on peripheral blood smear. Often associated with thrombocytopenia, renal failure, and/or neurological symptoms (e.g., in TTP/HUS). | Peripheral blood smear for schistocytes, platelet count, renal function tests, ADAMTS13 activity (for TTP). DAT is typically negative. |
| Hereditary Spherocytosis | Family history of anemia/jaundice/gallstones. Presence of spherocytes on peripheral blood smear, but DAT is negative. Splenomegaly is common. | Negative DAT. Osmotic fragility test, EMA binding test, flow cytometry for eosin-5-maleimide (EMA). |
| G6PD Deficiency | History of oxidant exposure (drugs like sulfa, antimalarials; fava beans) triggering hemolytic episodes. Presence of “bite cells” and Heinz bodies on peripheral smear during an acute crisis. | G6PD enzyme activity assay (should be done after acute hemolysis resolves). Negative DAT. |
| Paroxysmal Nocturnal Hemoglobinuria (PNH) | Episodes of dark urine (hemoglobinuria), especially in the morning. Associated with thrombosis (often in unusual sites) and bone marrow failure. | Flow cytometry for CD55 and CD59 on red blood cells (and often granulocytes). Negative DAT (though some cases may have weak positive due to complement). |
| Drug-Induced Immune Hemolytic Anemia (DIIHA) | Clear temporal relationship between drug exposure and onset of hemolysis. Drug-dependent antibodies can be detected. | Detailed drug history. Specialized laboratory tests for drug-dependent antibodies (not routinely available). DAT may be positive. |
| Hypersplenism | Significant splenomegaly (often palpable). Pancytopenia or bicytopenia may be present in addition to anemia. | Ultrasound/CT scan to assess spleen size. Bone marrow biopsy may be needed to rule out underlying cause of splenomegaly. DAT is negative. |
| Non-Hemolytic Anemias (e.g., Iron Deficiency, B12/Folate Deficiency, Anemia of Chronic Disease) | Lack of specific signs of hemolysis (e.g., no elevated indirect bilirubin, normal LDH, normal haptoglobin, no reticulocytosis). Red blood cell indices (MCV) can point to specific types (microcytic, macrocytic). | CBC with indices, iron studies, ferritin, Vitamin B12, folate, inflammatory markers (ESR, CRP). DAT is negative. |
| Secondary AIHA | The presence of underlying autoimmune disease (e.g., SLE, RA), lymphoproliferative disorder (e.g., CLL, lymphoma), or chronic infection (e.g., HIV, EBV). | Comprehensive workup for underlying conditions: ANA, RF, viral serologies, flow cytometry/biopsy for lymphoproliferative disorders. DAT is positive. |
Treatment and Management of AIHA
AIHA (Autoimmune Hemolytic Anemia) treatment focuses on suppressing antibody production or complement activation to prevent red blood cell destruction. The specific approach depends on the type of autoimmune hemolytic anemia (AIHA) (warm or cold).
Treatment Strategies
- Corticosteroids: The mainstay of therapy for both warm and cold autoimmune hemolytic anemia (AIHA). They suppress the immune system and antibody production.
- Rituximab: A B-cell depleting monoclonal antibody used in severe cases or when corticosteroids are not effective. It depletes B cells, which are responsible for antibody production.
- Splenectomy: May be considered as a last resort, particularly in warm autoimmune hemolytic anemia (AIHA), to remove the primary site of red blood cell destruction by macrophages in the spleen. However, it’s often avoided due to the increased risk of infections.
- Plasma Exchange/Immunoadsorption: These techniques remove antibodies and complement proteins from the blood circulation, providing temporary relief in severe cases.
- Supportive Care: This includes blood transfusions to manage severe anemia, hydration to prevent complications, and addressing any underlying conditions that might be triggering autoimmune hemolytic anemia (AIHA).
In some cases, treating the underlying condition causing autoimmune hemolytic anemia (AIHA) (e.g., infection, autoimmune disease) might be necessary to achieve long-term control.
Close monitoring is essential to assess the effectiveness of treatment and identify any potential side effects.
Comparison of Treatment Approaches
| Treatment | Warm AIHA | Cold Agglutinin Disease (CAD) |
| Corticosteroids (Prednisone) | First-line therapy | First-line therapy |
| Rituximab | Used in severe cases, non-responders to steroids | May be used earlier than in warm AIHA, especially if corticosteroids are ineffective |
| Splenectomy | Considered as a last resort | Rarely used due to potential for worsening symptoms |
| Plasma Exchange/ Immunoadsorption | May be used in severe cases with life-threatening hemolysis | Less commonly used compared to warm AIHA |
| Supportive Care | Crucial for managing anemia and preventing complications | Essential for managing anemia and addressing underlying triggers |
Prognosis of Autoimmune Hemolytic Anemia (AIHA)
The prognosis of AIHA (autoimmune hemolytic anemia) depends on several factors, including the type (warm or cold), severity, underlying cause (if any), and response to treatment.
Warm AIHA
Generally has a more variable prognosis compared to CAD.
- Favorable factors
- Prompt diagnosis and initiation of treatment
- Effective response to corticosteroids or rituximab
- Identification and management of any underlying cause (e.g., infection, autoimmune disease)
- Unfavorable factors
- Delayed diagnosis
- Severe anemia requiring frequent blood transfusions
- Relapses despite treatment
- Underlying health conditions that complicate treatment
CAD
Generally has a more favorable prognosis compared to warm AIHA.
- Favorable factors
- Less aggressive disease course in most cases
- May respond well to avoiding cold exposure (important for some patients)
- Treatment with corticosteroids or rituximab often effective
Long-term Management
In both warm and cold autoimmune hemolytic anemia (AIHA), long-term management may be necessary to control the condition and prevent complications. This might involve regular monitoring, maintaining immunosuppression with medications like low-dose corticosteroids or rituximab, and addressing any underlying triggers.
Frequently Asked Questions (FAQs)
How is AIHA related to thalassemia?
While uncommon, AIHA can develop in some individuals with thalassemia, particularly those with thalassemia major (the more severe form). The exact reasons for this association are not fully understood, but several factors might play a role:
- Increased immune system activation: Chronic anemia and repeated blood transfusions can put a strain on the immune system, potentially leading to the production of autoantibodies against red blood cells.
- Underlying genetic predisposition: Some individuals with thalassemia may have a genetic susceptibility to developing autoimmune disorders.
Symptoms of AIHA in Thalassemia
- Increased fatigue and weakness due to worsening anemia
- Pale skin
- Shortness of breath
- Jaundice (yellowing of the skin and eyes)
- Dark urine (in some cases)
What is the life expectancy with AIHA?
Recent studies suggest a potential decrease in life expectancy for AIHA patients compared to the general population. A Danish study found a median survival of 9.8 years for primary AIHA and 3.3 years for secondary AIHA.
Can AIHA be cured?
Autoimmune hemolytic anemia (AIHA) is often considered a chronic condition, meaning it can be a lifelong issue requiring ongoing management. Treatment strategies primarily focus on suppressing the immune system’s attack on red blood cells.
While not a guaranteed outcome, achieving long-term remission with minimal to no symptoms is possible for some patients, especially those with mild cases or specific types of autoimmune hemolytic anemia (AIHA) like cold agglutinin disease (CAD). Early diagnosis and prompt treatment are crucial for achieving remission and preventing complications.
Can AIHA cause low platelets?
AIHA (Autoimmune Hemolytic Anemia) typically affects red blood cells, and low platelets are not a characteristic finding of AIHA itself. However, there are a few scenarios where AIHA can be associated with low platelets:
- Evans Syndrome: This is a rare autoimmune disorder where the immune system attacks both red blood cells and platelets. This leads to a combination of anemia (low red blood cells) and thrombocytopenia (low platelets). If someone with autoimmune hemolytic anemia (AIHA) develops low platelets, it’s crucial to investigate for Evans Syndrome to ensure proper diagnosis and treatment.
- Underlying Cause of AIHA: In some cases, the underlying condition that triggers autoimmune hemolytic anemia (AIHA) might also affect platelets. For example, some viral infections or autoimmune diseases can cause both autoimmune hemolytic anemia (AIHA) and thrombocytopenia.
- Treatment Side Effects: Medications used to treat autoimmune hemolytic anemia (AIHA), such as corticosteroids, can sometimes have side effects that reduce platelet production, although this is uncommon.
What age does AIHA occur?
AIHA (Autoimmune Hemolytic Anemia) can occur at any age, but the risk factors and typical presentation can vary depending on the age group.
- Children: AIHA is relatively rare in children, but it can occur.
- Most Common Type: Cold agglutinin disease (CAD) is the most frequent form of AIHA in children.
- Trigger: Viral infections are a common trigger for CAD in children.
- Symptoms: Symptoms may be mild and resolve on their own without needing specific treatment for AIHA itself. However, managing the underlying infection is important.
- Prognosis: The prognosis for AIHA in children is generally good, especially with CAD.
- Adults: Autoimmune hemolytic anemia (AIHA) is more common in adults, with a slight peak in middle age (around 40-60 years old).
- Types: Both warm autoimmune hemolytic anemia (AIHA) and cold agglutinin disease (CAD) can occur in adults.
- Causes: Secondary autoimmune hemolytic anemia (AIHA) linked to underlying conditions like infections or autoimmune diseases becomes more prominent in adults. Primary AIHA also occurs in adults.
- Symptoms: Symptoms can vary depending on the severity of the condition. Early diagnosis and treatment are crucial.
- Older Adults: The risk of autoimmune hemolytic anemia (AIHA) might increase slightly again in older adults (above 65 years old).
- Considerations: Underlying medical conditions become even more common in this age group, potentially contributing to secondary autoimmune hemolytic anemia (AIHA).
- Diagnosis: It’s important to distinguish autoimmune hemolytic anemia (AIHA) from other causes of anemia in older adults.
Evans Syndrome, a rare autoimmune condition affecting both red blood cells and platelets, can occur at any age. Certain medications or blood transfusions can trigger autoimmune hemolytic anemia (AIHA) at any age.
What is the MCV level in autoimmune hemolytic anemia?
In autoimmune hemolytic anemia (AIHA), the MCV (Mean Corpuscular Volume) level is typically within the normal range (80-100 femtoliters [fL]). This is because autoimmune hemolytic anemia (AIHA) primarily affects the lifespan of red blood cells, not their size.
Is autoimmune hemolytic anemia inherited?
No, autoimmune hemolytic anemia (AIHA) itself is not directly inherited. It’s an autoimmune disorder, meaning the immune system malfunctions and attacks healthy tissues in the body, including red blood cells in the case of autoimmune hemolytic anemia (AIHA).
While genetics can play a role in susceptibility, autoimmune hemolytic anemia (AIHA) isn’t directly passed down from parents to children like a simple genetic trait.
Is autoimmune hemolytic anemia related to lymphoma?
Lymphoma, particularly Hodgkin’s lymphoma and non-Hodgkin lymphoma, can sometimes trigger autoimmune hemolytic anemia (AIHA) as a paraneoplastic syndrome. Here, the immune system becomes dysregulated due to the lymphoma, leading to autoantibody production against red blood cells.
Why does autoimmune hemolytic anemia caused spherocytes?
Autoimmune hemolytic anemia (AIHA) causes antibodies to bind to red blood cell membranes. The spleen, which normally filters blood, recognizes these antibody-coated cells as foreign. The spleen removes parts of the membrane to break down the “marked” cells. This loss of membrane surface area shrinks the red blood cell into a sphere (spherocyte), making it less functional and more susceptible to further destruction. This is more commonly seen in warm autoimmune hemolytic anemia (AIHA).
Disclaimer: This article is intended for informational purposes only and is specifically targeted towards medical students. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Jäger U, Barcellini W, Broome CM, Gertz MA, Hill A, Hill QA, Jilma B, Kuter DJ, Michel M, Montillo M, Röth A, Zeerleder SS, Berentsen S. Diagnosis and treatment of autoimmune hemolytic anemia in adults: Recommendations from the First International Consensus Meeting. Blood Rev. 2020 May;41:100648. doi: 10.1016/j.blre.2019.100648. Epub 2019 Dec 5. PMID: 31839434.
- Deng J, Zhou F, Wong CY, Huang E, Zheng E. Efficacy of therapeutic plasma exchange for treatment of autoimmune hemolytic anemia: A systematic review and meta-analysis of randomized controlled trials. J Clin Apher. 2020 Aug;35(4):294-306. doi: 10.1002/jca.21790. Epub 2020 May 8. PMID: 32384203.
- Swiecicki P, Hegerova LT, Gertz MA. Cold agglutinin disease. Blood 2013; 122(7):1114-1121.
- Go RS, Winters JL, Kay NE. How I treat autoimmune hemolytic anemia. Blood (2017) 129 (22): 2971–2979.
- Anita Hill, Quentin A. Hill. Autoimmune hemolytic anemia. Hematology Am Soc Hematol Educ Program (2018) 2018 (1): 382–389.
- https://www.msdmanuals.com/professional/hematology-and-oncology/anemias-caused-by-hemolysis/autoimmune-hemolytic-anemia
- Hansen DL, Möller S, Frederiksen H. Survival in autoimmune hemolytic anemia remains poor, results from a nationwide cohort with 37 years of follow-up. Eur J Haematol. 2022 Jul;109(1):10-20. doi: 10.1111/ejh.13764. Epub 2022 Mar 19. PMID: 35276014; PMCID: PMC9314695.