Key Takeaways
G6PD deficiency is the most common inherited enzyme defect of red blood cells, affecting an estimated 400 million people worldwide [4]. Most carriers are healthy until something triggers oxidative stress.
- G6PD deficiency symptoms & signs ▾: It is usually asymptomatic. Acute intravascular hemolytic anemia and hemoglobinuria can be seen during crises.
- Triggers ▾: Fava beans, certain antimalarials (primaquine, tafenoquine), dapsone, sulfonamide antibiotics, naphthalene (mothballs), and infections [2,3].
- G6PD deficiency tests ▾: Diagnosis uses a quantitative G6PD enzyme assay as the gold standard, supported by complete blood count, reticulocyte count, peripheral smear (Heinz bodies, bite cells), and bilirubin and LDH levels.
- G6PD deficiency treatment and management ▾: Mostly preventive: avoid triggers, treat infections early, and provide supportive care (hydration, transfusion, phototherapy in newborns) during a hemolytic crisis.
*Click ▾ for more information
What is G6PD deficiency?
G6PD deficiency is an inherited form of hemolytic anemia caused by a defective red blood cell enzyme. A red cell needs three things to do its job as an oxygen carrier: a healthy membrane, working metabolic pathways, and stable hemoglobin. Glucose-6-phosphate dehydrogenase sits inside one of those metabolic pathways — the pentose phosphate pathway (also called the hexose monophosphate shunt) — and protects the cell from oxidative damage [3].
Most people with G6PD deficiency feel completely well until a trigger (a fava bean meal, a course of antimalarial drugs, or an infection) sets off a sudden episode of red cell breakdown. Knowing what to avoid is what keeps someone safe.
Inheritance and Epidemiology
The G6PD gene sits on the long arm of the X chromosome, at band Xq28. Inheritance is X-linked recessive. Males have only one X chromosome, so a single altered copy produces full deficiency. Females have two X chromosomes and are usually carriers — but carrier status is not the same as "unaffected." Because of X-inactivation, each cell randomly silences one X chromosome, leaving heterozygous females with a mosaic of G6PD-normal and G6PD-deficient red cells. A meaningful proportion of these women can still hemolyze when exposed to strong oxidants like primaquine or tafenoquine [5].
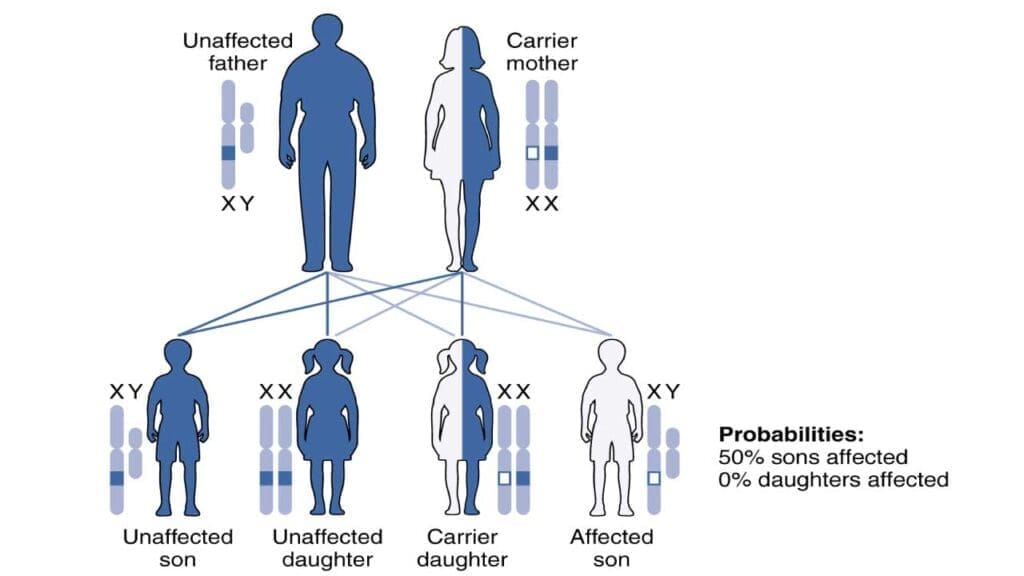
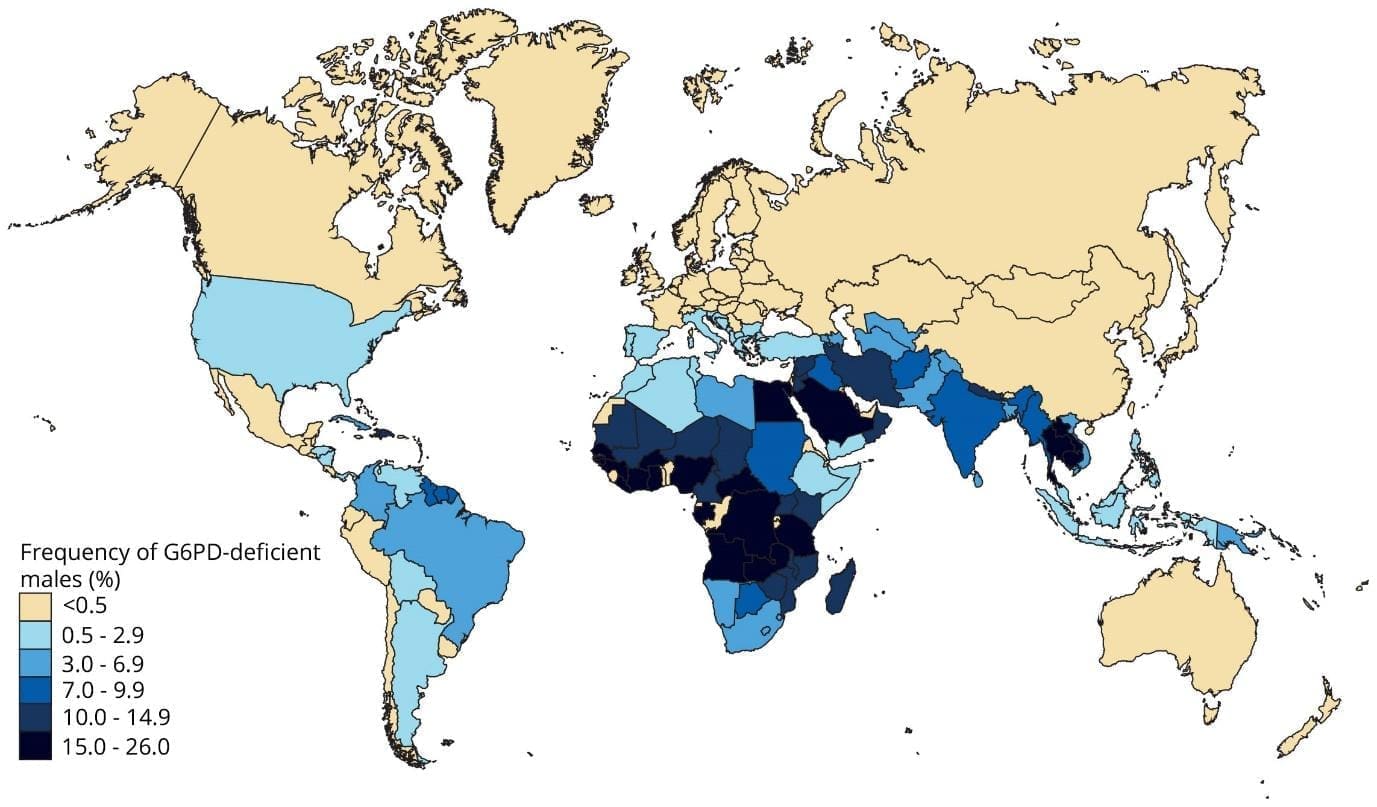
G6PD deficiency is the most common red cell enzymopathy in the world. The widely cited geostatistical estimate puts the average allele frequency at roughly 8% across malaria-endemic countries, with about 400 million people affected globally [4]. Prevalence is highest in sub-Saharan Africa, the Mediterranean basin, the Middle East, and South and Southeast Asia. The variant matters as much as the prevalence: the African A− variant is generally milder, while the Mediterranean variant tends to cause more severe hemolysis [3].
The geographic overlap with malaria is not a coincidence. G6PD deficiency offers a small protective advantage against severe Plasmodium falciparum malaria, which is why the deficiency persists at high frequency in these regions [3].
Why G6PD Matters Inside the Red Cell
G6PD catalyzes the first and rate-limiting step of the pentose phosphate pathway. It uses glucose-6-phosphate to convert NADP⁺ into its reduced form, NADPH.
NADPH is the red cell's main antioxidant currency. It powers glutathione reductase, which keeps glutathione in its reduced form (GSH). Glutathione peroxidase then uses GSH to neutralize hydrogen peroxide, turning it into water. So G6PD does not detoxify peroxide directly. It generates the NADPH that makes the whole defense system work [3].
Mature red cells are particularly vulnerable. They cannot make new proteins or repair damage, so once their antioxidant supply runs out, oxidative damage accumulates fast.
Pathophysiology of a Hemolytic Crisis
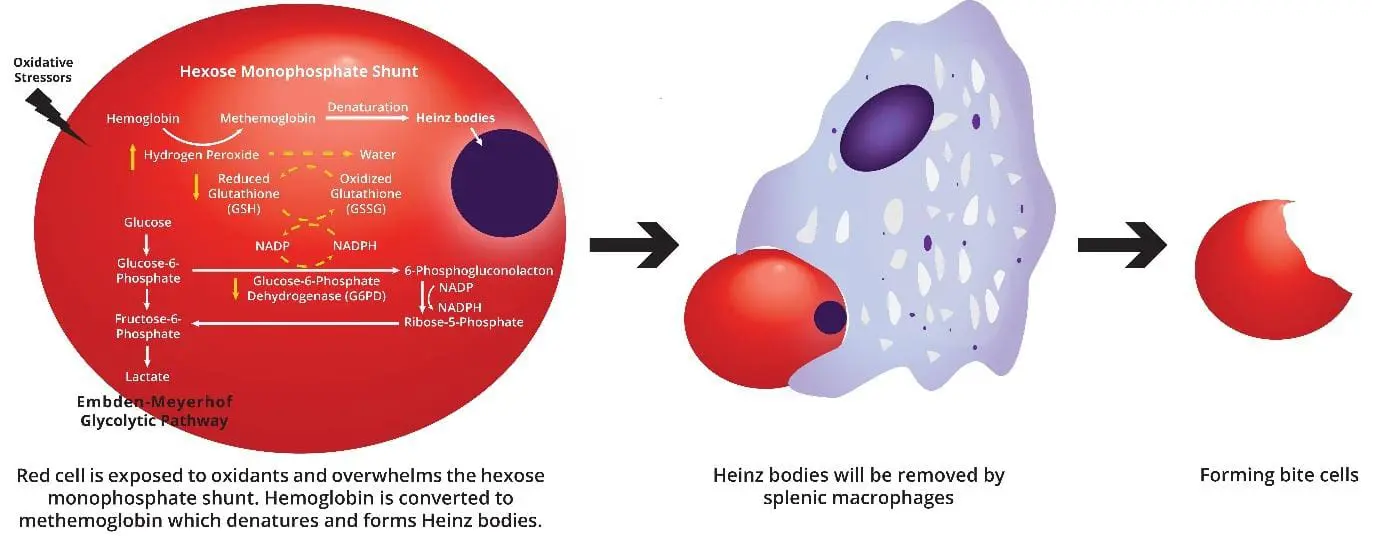
When a G6PD-deficient red cell meets an oxidant (a drug, an infection by-product, or a fava bean compound), it cannot regenerate enough NADPH. Reduced glutathione runs out. Hydrogen peroxide and other reactive species attack hemoglobin and the membrane. Damaged hemoglobin precipitates inside the cell as Heinz bodies (clumps of denatured protein).
The spleen recognizes these damaged cells. Splenic macrophages either destroy the cell entirely or "bite off" the Heinz body, leaving the characteristic bite cell on a peripheral smear. The result is a mixture of intravascular and extravascular hemolysis: dark urine from free hemoglobin, jaundice from bilirubin, and falling hemoglobin levels [3,6].
What Triggers a Hemolytic Crisis?
A G6PD-deficient red cell handles normal day-to-day metabolism fine. The problem is acute oxidative stress. The 2022 expanded CPIC pharmacogenetics guideline reviewed the evidence for 48 drugs and grouped them by risk [2]. That framework, plus dietary and infectious triggers, gives the practical list below.
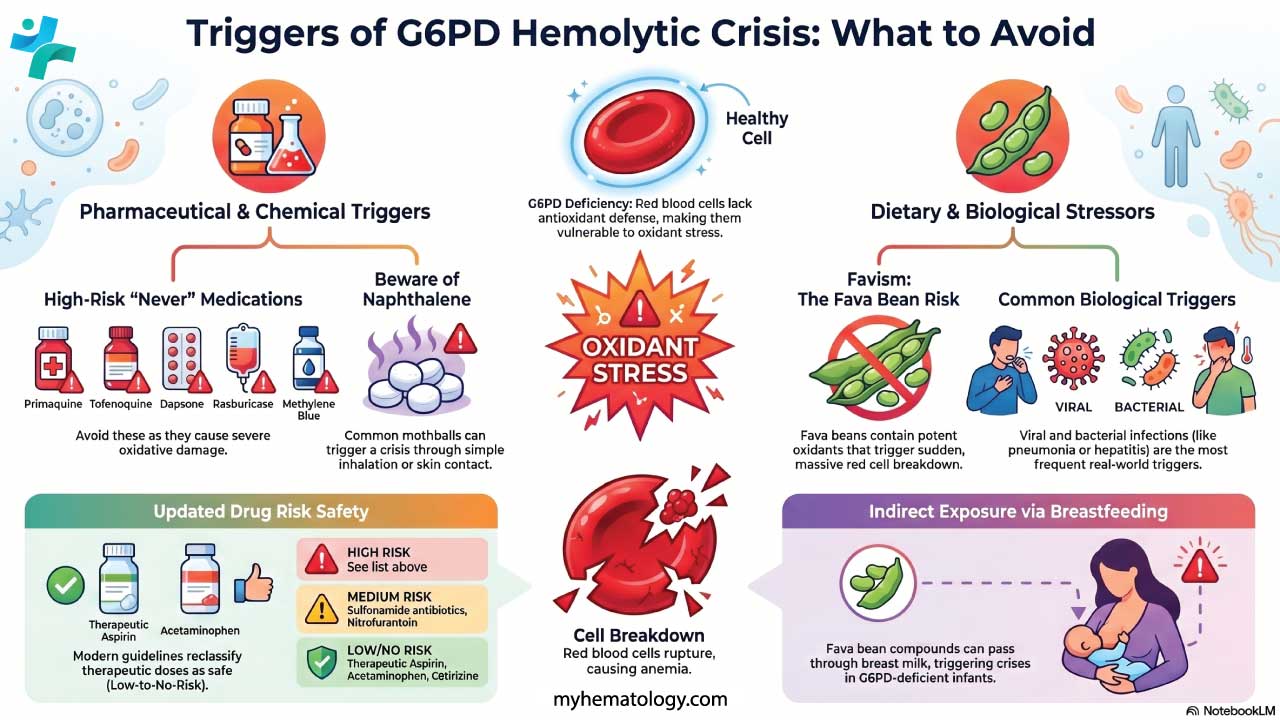
High-Risk Drugs (Avoid)
- Primaquine and tafenoquine (antimalarials targeting dormant liver-stage P. vivax)
- Dapsone (used for leprosy, Pneumocystis jirovecii prophylaxis, and some skin conditions)
- Rasburicase (used for tumor lysis syndrome)
- Methylene blue
- Pegloticase (a recombinant uricase used for severe, treatment-refractory gout) [9]
- Toluidine blue (a dye used in various medical diagnostics and procedures)
- Phenazopyridine (a common over-the-counter and prescription urinary tract analgesic)
Medium-Risk Drugs (Use With Caution)
- Sulfonamide antibiotics such as sulfamethoxazole
- Nitrofurantoin
- Some other antimalarials including chloroquine in certain settings
Low-to-No-Risk Drugs (Standard Use Is Safe)
- Therapeutic-dose aspirin and most common NSAIDs
- Acetaminophen / paracetamol at therapeutic doses
- Cetirizine and most second-generation antihistamines
This stratification matters because outdated lists still circulating online include many drugs that the 2022 guideline reclassified as low-risk. Students should learn the framework, not memorize a flat list.
Foods
The classic dietary trigger is fava beans (broad beans), which contain divicine and isouramil — both potent oxidants. The resulting acute hemolysis is called favism. Other foods (blueberries, tonic water, some traditional remedies) are mentioned in the older literature, but the evidence is much weaker.
Breastfeeding mothers of G6PD-deficient infants must also strictly avoid fava beans, as the hemolysis-inducing compounds can readily pass through breast milk and trigger severe neonatal jaundice or a hemolytic crisis in exclusively breastfed infants [7].
Infections
Bacterial and viral infections including hepatitis, pneumonia, and typhoid fever are among the most common triggers in real-world practice [3,6]. Inflammation generates reactive oxygen species, and fever raises metabolic demand. This is why prompt treatment of infection is part of G6PD management.
Other Oxidative Stressors
- Naphthalene (mothballs), through inhalation or skin contact
- Diabetic ketoacidosis during rapid correction
- Severe physical or emotional stress (rare but reported)
Methylene Blue Danger in G6PD Deficient Patients
Methylene blue is the standard antidote for methemoglobinemia in healthy patients. In G6PD-deficient patients it does the opposite — it relies on NADPH to do its work, and giving it to someone short on NADPH can worsen oxidative damage.
G6PD Deficiency Symptoms
Most of the time, there are none. G6PD deficiency is usually asymptomatic between triggers, and many people never know they have it.
When a hemolytic crisis does happen, symptoms appear within hours to a couple of days of the trigger:
- Pallor (pale skin and mucous membranes) from falling red cell mass
- Jaundice (yellowing of the skin and the whites of the eyes) from bilirubin released by destroyed cells
- Dark, tea-colored, or red-brown urine from hemoglobinuria
- Fatigue, weakness, and shortness of breath from reduced oxygen delivery
- Tachycardia as the heart compensates
- Back or abdominal pain during severe hemolysis
- Fever, especially when an infection is the trigger
- Splenomegaly (an enlarged spleen) that may be tender
Newborns

G6PD deficiency is a leading cause of neonatal jaundice in many parts of the world. Bilirubin can rise quickly and, if untreated, may cross the blood-brain barrier and cause kernicterus, a permanent brain injury [3,6]. Warning signs include deep or rapidly-deepening jaundice, lethargy, poor feeding, a high-pitched cry, abnormal muscle tone, arching of the back, and seizures.
Long-Term Complications
Most people with G6PD deficiency live normal lives. Repeated severe crises can occasionally lead to bilirubin gallstones, acute kidney injury, or in the rare class A variants — chronic non-spherocytic hemolytic anemia (CNSHA), where hemolysis persists even without obvious triggers [1,3].
G6PD Variants and How They Affect Severity
Different mutations in the G6PD gene produce different levels of residual enzyme activity. WHO updated the G6PD variant classification system (endorsed by the WHO Malaria Policy Advisory Group in March 2022, and formally published and integrated into guidelines in 2024) [1]. The new system has four classes.
G6PD Deficiency
| Class | Enzyme Activity | Clinical Presentation | |
|---|---|---|---|
| A Severe | < 20% of normal | Chronic non-spherocytic hemolytic anemia (CNSHA) — hemolysis occurs constitutively, even in the absence of an exogenous trigger. | |
| B Moderate | < 45% of normal | Acute hemolysis triggered by oxidative stressors — including drugs, infections, or fava bean ingestion. Asymptomatic at baseline. | |
| C Normal | 60 – 150% of normal | No clinically significant hemolysis. Enzyme activity within or above the reference range. | |
| U Uncertain | Any level | Variant of uncertain clinical significance — insufficient evidence to classify clinical risk based on current data. |
The biggest change was merging the old classes II and III into a single class B [1]. Studies showed that the 10% activity cutoff between them did not actually predict clinical risk well, because individual enzyme activity varied widely within both classes. Class B now includes the most common polymorphic variants worldwide, including African A−, Mediterranean, and Mahidol — all of which carry a real risk of severe acute hemolysis and neonatal jaundice [1].
The Older WHO Classification (Class I–V)
The five-class system (severe → normal) is what older textbooks still use but the four-class system is the current standard.
How is G6PD Deficiency Diagnosed?
Investigation depends on whether the patient is in the middle of a hemolytic episode or being screened in advance.
Tests During Acute Hemolysis
A complete blood count typically shows anemia with a low hemoglobin and hematocrit. The reticulocyte count rises as the bone marrow compensates. A peripheral blood smear shows bite cells, blister cells, polychromatic cells, and microspherocytes. Heinz bodies are visible with a supravital stain such as crystal violet or brilliant cresyl blue.
Markers of hemolysis follow a predictable pattern:
- Unconjugated (indirect) bilirubin: elevated — released from destroyed cells
- Lactate dehydrogenase (LDH): elevated — a general cell-damage marker
- Haptoglobin: low — consumed as it binds free hemoglobin
- Urinalysis: hemoglobinuria during severe intravascular hemolysis
G6PD Enzyme Activity Tests
These directly measure how much enzyme is working in the red cells. Timing matters: testing during or just after a hemolytic crisis can give falsely normal results, because young reticulocytes have higher G6PD activity than mature red cells. Retest two to three months after recovery for an accurate result.
- Fluorescent spot test (FST): A widely used qualitative screen. NADPH made by G6PD fluoresces under UV light; reduced fluorescence suggests deficiency [3,6].
- Quantitative spectrophotometric assay: The gold standard. It measures the rate of NADPH formation precisely.
- Quantitative point-of-care tests: Newer near-patient devices provide a numerical activity result in minutes and have high sensitivity and specificity for diagnosing G6PD deficiency [10]. Most notably, the SD Biosensor STANDARD G6PD System recently received WHO prequalification, establishing it as the internationally recognized tool to confirm G6PD activity before administering primaquine or tafenoquine in malaria-endemic settings.
Molecular Genetic Testing
DNA analysis identifies the specific G6PD variant. It is most useful for:
- Confirming the diagnosis when phenotypic results are unclear
- Identifying heterozygous female carriers (where enzyme activity may be intermediate)
- Newborn testing, when reticulocytosis or transfusion confounds enzyme assays
- Genetic counseling
Newborn and Pre-Drug Screening
Many countries with high G6PD prevalence — including Malaysia, Singapore, parts of southern Europe, and several US states — run routine newborn screening programs. The goal is to catch affected infants before severe jaundice develops [3].
Pre-drug screening has become more important with the wider use of tafenoquine. Because tafenoquine is given as a single dose, a clinician cannot stop the drug if hemolysis begins. Current WHO guidance is to confirm G6PD activity above the relevant threshold (typically ≥70% for full primaquine radical cure and ≥30% for single-dose tafenoquine) before treatment [1].
Diagnostic Interference in Diabetes
G6PD deficiency can artificially lower hemoglobin A1c (HbA1c) levels. Because G6PD-deficient red blood cells undergo subclinical oxidative stress and have a shortened lifespan, there is less time for hemoglobin to become glycated. This discrepancy can mask abnormal glucose tolerance and lead to a delayed diagnosis of Type 2 Diabetes, particularly in demographics with high genetic prevalence. For patients with known G6PD deficiency, clinicians should interpret HbA1c cautiously and utilize alternative glycemic screening methods, such as fasting plasma glucose or continuous glucose monitoring, to prevent delayed care.

How is G6PD Deficiency Treated?
There is no cure. Management of G6PD deficiency rests on two pillars: preventing crises and supportive care when one happens [3,6].
Prevention (the Main Strategy)
- Give the patient a written list of drugs to avoid, ideally based on the 2022 CPIC guideline [2]. Update it whenever they start a new medication.
- Avoid fava beans in any form, including dried, fresh, or as flour.
- Treat infections promptly. Many crises start with a febrile illness.
- Avoid naphthalene (mothballs and some industrial chemicals).
- Tell every prescriber, dentist, and surgeon about the deficiency before any new drug or procedure.
Supportive Care During a Hemolytic Crisis
- Stop the trigger. This is the single most important step.
- Hydrate with intravenous fluids if hemoglobinuria is significant, to reduce the risk of acute kidney injury.
- Transfuse packed red blood cells if the anemia is severe enough to compromise oxygen delivery.
- Donor blood considerations: When transfusions are necessary, be aware that transfusing red blood cells from a G6PD-deficient donor results in decreased in vivo recovery and reduced survival of the transfused cells [8]. This is especially critical for patients requiring chronic transfusions (such as those with sickle cell disease), making the screening of donor blood for G6PD status highly advisable in high-prevalence areas.
- Treat the underlying infection, if there is one.
- Monitor hemoglobin, bilirubin, renal function, and urine output until the patient stabilizes.
Treating Newborn Jaundice
- Phototherapy with blue light converts unconjugated bilirubin into a water-soluble form the baby can excrete.
- Exchange transfusion is reserved for very high bilirubin levels and a real risk of kernicterus.
When to Consider Splenectomy
Splenectomy is rarely needed and is reserved for the small subset of patients with class A (chronic) variants and severe ongoing hemolysis [1,3]. For everyone else, trigger avoidance is the answer.
Frequently Asked Questions (FAQs)
Is G6PD deficiency a form of anemia?
Not exactly. G6PD deficiency is a genetic enzyme defect that puts red cells at risk of breaking down under oxidative stress. The anemia only appears during a hemolytic episode triggered by a drug, infection, or fava beans. Many people with G6PD deficiency have normal blood counts most of their lives.
Can women have G6PD deficiency?
Yes. The gene is on the X chromosome, so men are typically more severely affected. But heterozygous female carriers can still hemolyze, especially when given strong oxidant drugs like primaquine or tafenoquine. This happens because of X-inactivation, which leaves a mix of G6PD-deficient and G6PD-normal red cells in their blood [5].
What foods and medicines should someone with G6PD deficiency avoid?
The clearest avoidances are fava beans, the antimalarials primaquine and tafenoquine, dapsone, rasburicase, methylene blue, and naphthalene (mothballs). The 2022 CPIC pharmacogenetics guideline classifies these as high-risk [2]. Sulfonamide antibiotics fall in a medium-risk group. Therapeutic-dose aspirin, acetaminophen, and cetirizine are now considered low-to-no risk. Patients should always check medications against an up-to-date G6PD-safe list and tell every prescriber about their deficiency.
Why is the G6PD test important before malaria treatment?
Modern antimalarials primaquine and tafenoquine clear dormant Plasmodium vivax from the liver. Both can cause severe hemolysis in G6PD-deficient people. Tafenoquine is given as a single dose, so once it is in the body, it cannot be stopped. WHO recommends confirming G6PD activity before either drug, ideally with a quantitative point-of-care test [1].
Will my child outgrow G6PD deficiency?
No. G6PD deficiency is a lifelong genetic condition. The good news is that most affected children grow up healthy as long as triggers are avoided. The most important early concern is severe newborn jaundice, which is treatable with phototherapy and, if needed, exchange transfusion.
Is there a cure for G6PD deficiency?
Not currently. Management is built on prevention — avoiding triggers, treating infections promptly, and getting prompt care during a hemolytic episode. Research into gene therapy and small-molecule activators of the G6PD enzyme is ongoing but not yet available in clinical practice.
Disclaimer: This article is intended for educational and informational purposes only. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Luzzatto, L., Bancone, G., Dugué, P. A., Jiang, W., Minucci, A., Nannelli, C., Pfeffer, D., Prchal, J., Sirdah, M., Sodeinde, O., Vulliamy, T., Wanachiwanawin, W., Cunningham, J., & Bosman, A. (2024). New WHO classification of genetic variants causing G6PD deficiency. Bulletin of the World Health Organization, 102(8), 615–617. https://doi.org/10.2471/BLT.23.291224
- Gammal, R. S., Pirmohamed, M., Somogyi, A. A., Morris, S. A., Formea, C. M., Elchynski, A. L., Oshikoya, K. A., McLeod, H. L., Haidar, C. E., Whirl-Carrillo, M., Klein, T. E., Caudle, K. E., & Relling, M. V. (2023). Expanded Clinical Pharmacogenetics Implementation Consortium Guideline for Medication Use in the Context of G6PD Genotype. Clinical pharmacology and therapeutics, 113(5), 973–985. https://doi.org/10.1002/cpt.2735
- Luzzatto, L., Ally, M., & Notaro, R. (2020). Glucose-6-phosphate dehydrogenase deficiency. Blood, 136(11), 1225–1240. https://doi.org/10.1182/blood.2019000944
- Howes, R. E., Piel, F. B., Patil, A. P., Nyangiri, O. A., Gething, P. W., Dewi, M., Hogg, M. M., Battle, K. E., Padilla, C. D., Baird, J. K., & Hay, S. I. (2012). G6PD deficiency prevalence and estimates of affected populations in malaria endemic countries: a geostatistical model-based map. PLoS medicine, 9(11), e1001339. https://doi.org/10.1371/journal.pmed.1001339
- Chu, C. S., Bancone, G., Nosten, F., White, N. J., & Luzzatto, L. (2018). Primaquine-induced haemolysis in females heterozygous for G6PD deficiency. Malaria journal, 17(1), 101. https://doi.org/10.1186/s12936-018-2248-y
- Mak GK, Shah M. Glucose-6-Phosphate Dehydrogenase Deficiency. [Updated 2025 Nov 30]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2026 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK470315/
- Beretta, A., Manuelli, M., & Cena, H. (2023). Favism: Clinical Features at Different Ages. Nutrients, 15(2), 343. https://doi.org/10.3390/nu15020343
- Francis, R. O., Jhang, J. S., Pham, H. P., Hod, E. A., Zimring, J. C., & Spitalnik, S. L. (2013). Glucose-6-phosphate dehydrogenase deficiency in transfusion medicine: the unknown risks. Vox sanguinis, 105(4), 271–282. https://doi.org/10.1111/vox.12068
- McDonagh, E. M., Thorn, C. F., Callaghan, J. T., Altman, R. B., & Klein, T. E. (2014). PharmGKB summary: uric acid-lowering drugs pathway, pharmacodynamics. Pharmacogenetics and genomics, 24(9), 464–476. https://doi.org/10.1097/FPC.0000000000000058
- Pal, S., Bansil, P., Bancone, G., Hrutkay, S., Kahn, M., Gornsawun, G., Penpitchaporn, P., Chu, C. S., Nosten, F., & Domingo, G. J. (2019). Evaluation of a Novel Quantitative Test for Glucose-6-Phosphate Dehydrogenase Deficiency: Bringing Quantitative Testing for Glucose-6-Phosphate Dehydrogenase Deficiency Closer to the Patient. The American journal of tropical medicine and hygiene, 100(1), 213–221. https://doi.org/10.4269/ajtmh.18-0612