Key Takeaways
Sickle cell disease (SCD) is a group of inherited blood disorders, not a single condition. All forms involve hemoglobin S (HbS), produced by a mutation in codon 6 of the β-globin gene that swaps glutamic acid for valine.
- Pathophysiology ▾: HbS polymerizes when oxygen is low, distorting red cells into stiff sickle shapes that block small blood vessels (vaso-occlusion) and break down early (chronic hemolysis).
- Signs and symptoms ▾: The shared clinical picture includes pain crises, anemia, and a high risk of stroke, acute chest syndrome, and infection, though the frequency and pattern vary by genotype.
- Diagnosis ▾: Diagnosis confirms the presence, quantity, and combination of hemoglobins using HPLC or electrophoresis, with DNA testing for ambiguous or prenatal cases.
- Treatment and management ▾: Hydroxyurea is the foundation of disease-modifying treatment for sickle cell disease.
*Click ▾ for more information
What does a red blood cell actually do?
A red blood cell is essentially a sealed bag of hemoglobin. Its job is to carry oxygen from the lungs to every tissue and bring carbon dioxide back. To do that well, it needs a flexible membrane, working metabolism, and structurally normal hemoglobin. A single mature red cell holds roughly 250–280 million hemoglobin molecules.
Hemoglobin is a tetramer: two α-like and two β-like globin chains, each cradling a heme group with one iron atom. Each iron binds one oxygen molecule. The α-globin genes sit on chromosome 16 and the β-globin genes on chromosome 11. Different combinations are made at different life stages. Fetal hemoglobin (HbF) binds oxygen tightly to pull it across the placenta. After birth, production switches over the first six months to adult hemoglobin (HbA), which releases oxygen more readily to tissues.
When the genes for these chains are mutated, the result is a hemoglobinopathy.
What are hemoglobinopathies?
Hemoglobinopathies are inherited mutations affecting the globin genes. They split into two groups. Structural hemoglobinopathies produce a hemoglobin molecule with a faulty shape, such as HbS or HbC. Quantitative hemoglobinopathies, the thalassemias, produce too little of one or more globin chains. Sickle cell disease sits mostly in the first group, but some of its forms combine a structural and a quantitative defect, which is why HbS/β-thalassemia exists.
What is sickle cell disease?
Sickle cell disease is an umbrella term for the inherited conditions in which red cells sickle because they carry hemoglobin S. The shared root cause is the same single DNA change: in codon 6 of the β-globin gene, glutamic acid (a hydrophilic amino acid) is replaced by valine (a hydrophobic one). This swap makes the hemoglobin polymerize and the red cell deform when oxygen drops.
What separates the forms of SCD is the second β-globin gene a person inherits.
- Sickle cell anemia (HbSS): two sickle genes. The most common and most severe form.
- HbSC disease: one sickle gene and one hemoglobin C gene. Generally milder anemia, but high rates of retinopathy and avascular necrosis.
- HbS/β⁰-thalassemia: one sickle gene and a β-thalassemia gene that makes no β-globin. Clinically resembles HbSS.
- HbS/β⁺-thalassemia: one sickle gene and a β-thalassemia gene that makes some normal β-globin. Usually the mildest of the major forms.
- Rare combinations: HbSD-Punjab, HbSE, and others.
| Genotype | Inheritance | Typical Clinical Picture |
|---|---|---|
|
HbAA
|
Two normal β genes | Healthy |
|
HbAS
|
One sickle gene (trait) | Usually asymptomatic; carrier |
|
HbSS
|
Two sickle genes | Sickle cell anemia; most severe |
|
HbSC
|
One sickle, one HbC gene | Milder anemia; high rates of retinopathy and avascular necrosis |
|
HbS/β⁰-thal
|
One sickle, one β⁰ gene | Clinically resembles HbSS |
|
HbS/β⁺-thal
|
One sickle, one β⁺ gene | Usually the mildest major form |
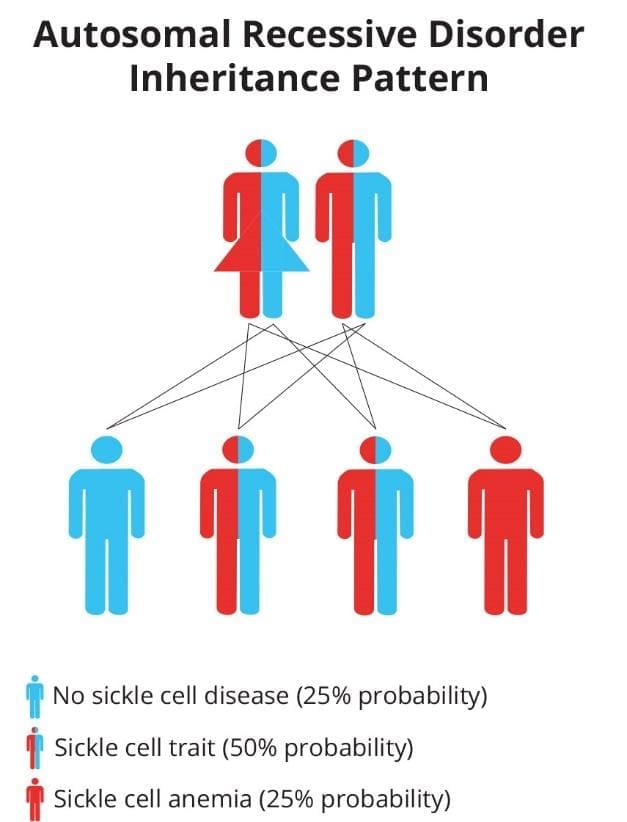
All of these are inherited in an autosomal recessive pattern, meaning a child needs two abnormal β-globin genes to have the disease. Inheriting one sickle gene and one normal gene gives sickle cell trait (HbAS), which is usually symptom-free and is a carrier state, not a form of the disease.
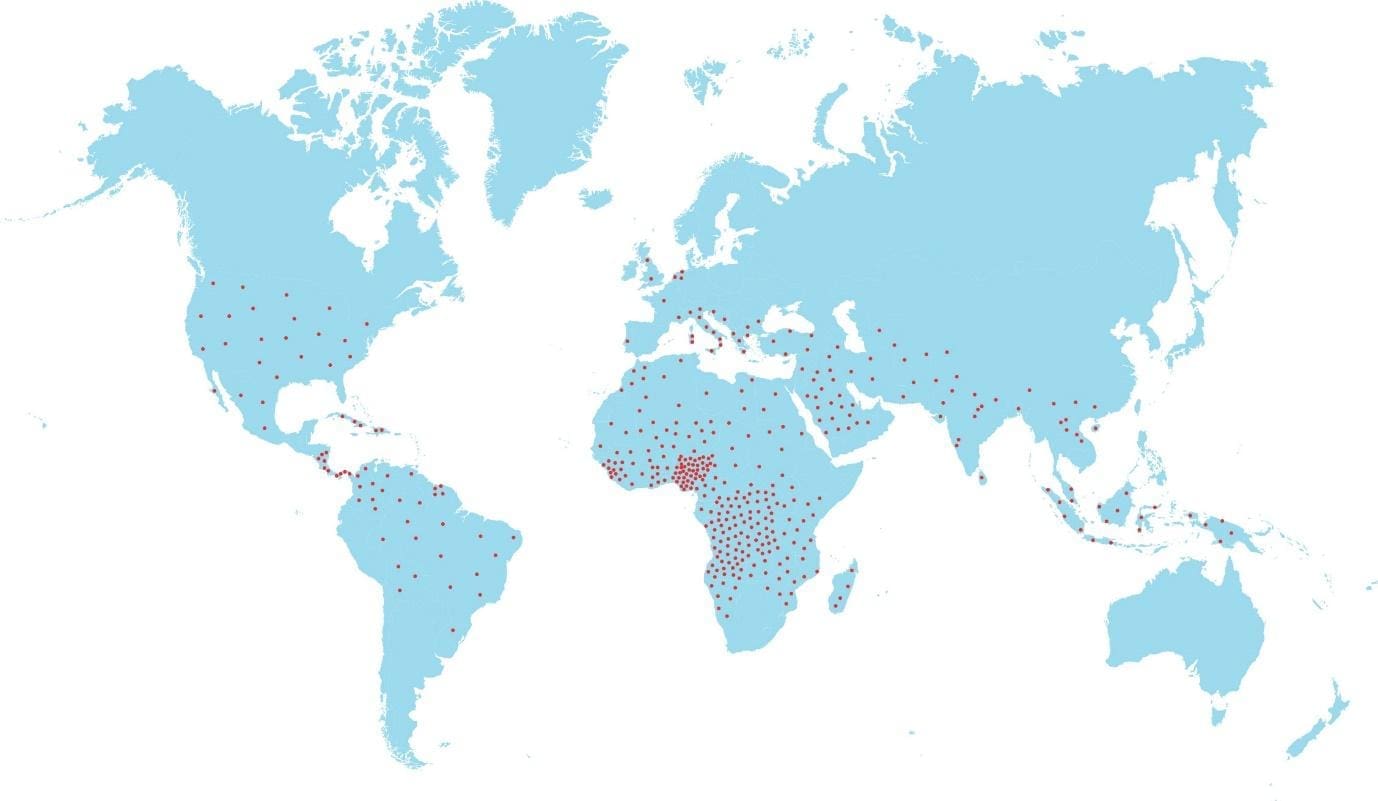
Where is sickle cell disease common?
Sickle cell disease clusters in sub-Saharan Africa, parts of the Mediterranean, the Middle East, and South Asia, mirroring the historical distribution of malaria. One copy of the sickle gene offers some protection against severe Plasmodium falciparum malaria, which is why the mutation persisted across generations [8]. HbC, which combines with HbS to cause HbSC disease, is concentrated in West Africa.
How sickle cell disease causes harm
Two intertwined processes drive nearly every complication: vaso-occlusion and chronic hemolysis. Both follow from the same molecular event, though their balance shifts between genotypes.
When HbS gives up its oxygen, the molecules link into long fibers (called tactoids) inside the red cell. Brief deoxygenation causes reversible sickling. Repeated cycles damage the membrane, expose sticky adhesion molecules, and lock some cells into permanent sickle shapes. These stiff, sticky cells then misbehave in two ways.
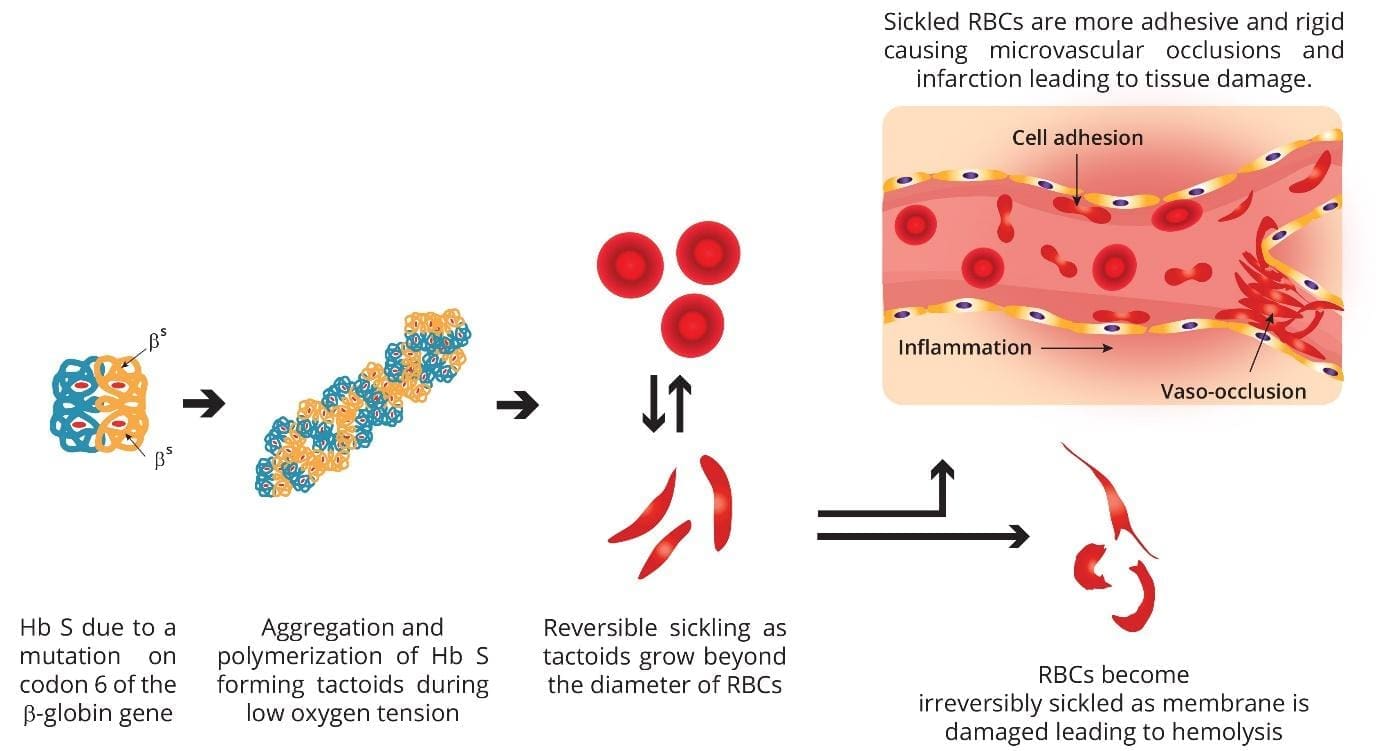
The degree of hemolysis varies by genotype. HbSS and HbS/β⁰-thalassemia hemolyze heavily, so anemia is prominent. HbSC and HbS/β⁺-thalassemia hemolyze less, so these patients often have higher baseline hemoglobin and milder anemia, even though they remain at risk for vaso-occlusive complications.
Free hemoglobin released during hemolysis scavenges nitric oxide (NO), the molecule that keeps blood vessels relaxed. Loss of NO leads to chronic vasoconstriction and contributes to pulmonary hypertension, a major cause of death in adults with SCD [4,7].
End-organ damage adds up
Years of vaso-occlusion and inflammation cause permanent injury: stroke, acute chest syndrome, kidney disease, avascular necrosis of the hip and shoulder, retinopathy, and gallstones from chronic bilirubin overload.
The spleen illustrates how genotype changes the story. In HbSS, the spleen is usually scarred into uselessness by early childhood, a state called autosplenectomy. In HbSC and HbS/β⁺-thalassemia, the spleen often survives into adulthood and can even enlarge, which keeps the risk of acute splenic sequestration alive much later in life than in HbSS.
Signs and symptoms
Symptoms appear after the HbF-to-HbA switch is well under way, typically between 6 and 12 months of age. The pattern shifts with age, and milder genotypes may not declare themselves until later childhood or adulthood.
Early Childhood
Acute crises
A vaso-occlusive crisis is the hallmark event across all genotypes. Pain is sudden, often in the bones of the back, chest, arms, or legs, and can last hours to weeks.
Acute chest syndrome (ACS) looks like pneumonia: chest pain, fever, cough, and shortness of breath, with a new lung infiltrate on chest X-ray. It is a leading cause of death in adults with SCD and demands rapid treatment.
Splenic sequestration traps blood in the spleen, which swells dramatically as hemoglobin drops fast. In HbSS it strikes young children before the spleen scars; in HbSC and HbS/β⁺-thalassemia it can occur in older children and adults because the spleen persists. Either way it is a true emergency.
Aplastic crisis is a temporary shutdown of red-cell production by the bone marrow, almost always triggered by parvovirus B19. The reticulocyte count falls and anemia worsens sharply.
Stroke is most common in children aged 2–9 with the severe genotypes (HbSS, HbS/β⁰-thalassemia). Sudden weakness, slurred speech, facial droop, or severe headache should prompt immediate evaluation.
Chronic findings
Pallor, fatigue, and jaundice (yellow skin and sclerae from elevated bilirubin) reflect ongoing hemolysis and are most prominent in the severe genotypes. Adults often develop chronic joint pain from avascular necrosis, leg ulcers, retinopathy, kidney disease, and pulmonary hypertension.
Early investigation of persistent joint pain requires an MRI, as plain X-rays often appear completely normal in the early, reversible stages of avascular necrosis [18]. Notably, proliferative sickle retinopathy is more common in HbSC disease than in HbSS, a useful exam point. Priapism, a prolonged painful erection, can occur at any age and is itself a vaso-occlusive event.
Sickle cell trait: a carrier state, not a disease
People with one sickle gene (HbAS) have sickle cell trait. Most are completely asymptomatic. They carry the gene and can pass it on; if both parents have trait, each pregnancy carries a 25% chance of producing a child with sickle cell disease. Trait offers measurable protection against severe malaria, which explains the geography. It can occasionally cause problems under extreme conditions: dehydration, very high altitude, or intense exertion. It is not a form of sickle cell disease and should not be described as a "mild" version of it.
How is sickle cell disease diagnosed?
Investigation has two layers: detecting HbS, then defining the exact genotype. Pinning down the genotype matters more for SCD than for HbSS alone, because it predicts the likely course and tailors monitoring.
Screening
- Newborn screening (NBS) is mandatory in all US states and many other high-income countries, allowing infants to start prophylactic penicillin within weeks of birth. Universal NBS is still being scaled up in much of sub-Saharan Africa and South Asia, where most affected children are born.
- Hemoglobin solubility (sickling) test is a rapid, cheap screen. Deoxygenated HbS is poorly soluble, so the solution turns cloudy. It detects the presence of HbS but cannot distinguish among genotypes; it cannot tell HbSS from HbAS or HbSC. It can be falsely negative in infants with high HbF or in severely anemic patients. It is not a definitive test.
| Test | How it works | What it tells you |
|---|---|---|
| Hemoglobin Electrophoresis | Separates hemoglobin variants by electrical charge |
Identifies hemoglobin variants; less precise for quantification. |
| Separates hemoglobin by chemical properties on a column; quantifies each fraction |
Quantifies hemoglobin fractions and distinguishes key genotypes. |
|
| DNA Analysis (PCR) | Detects specific HBB gene mutations at the molecular level |
Resolves ambiguous results; used for prenatal diagnosis. |
Routine and supportive tests
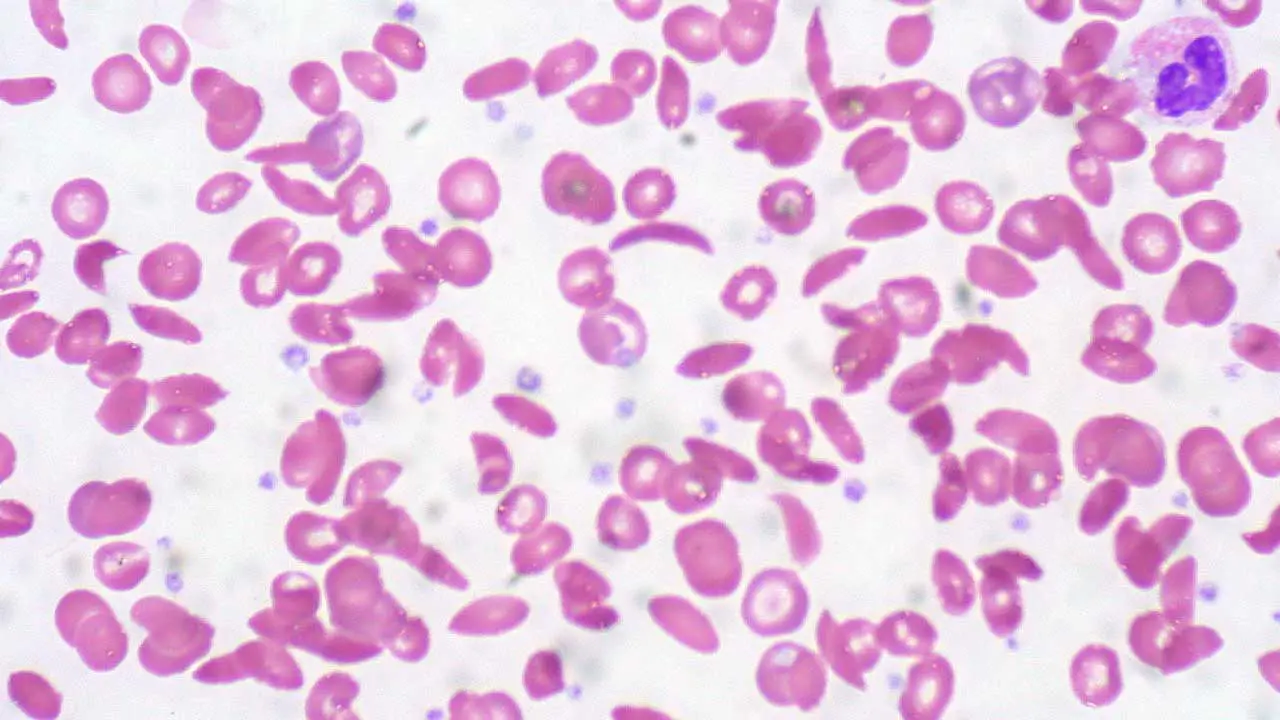
A complete blood count (CBC) typically shows anemia (marked in HbSS, milder in HbSC) with a high reticulocyte count from a marrow working overtime, an elevated white cell count from chronic inflammation, and sometimes a low mean corpuscular volume in the thalassemia combinations. The peripheral blood smear shows sickled cells, target cells, and Howell-Jolly bodies; in HbSC, distinctive cells with crystalline inclusions may appear.
Transcranial Doppler (TCD) ultrasonography screens for stroke risk in children aged 2–16 with the severe genotypes. High blood-flow velocity predicts stroke and triggers preventive transfusion programs.
How is sickle cell anemia treated?
Modern care has three goals: prevent crises, slow chronic organ damage, and where possible, cure the disease. The principles are shared across genotypes, though milder forms may need less intensive management.
Daily disease-modifying therapy
Hydroxyurea (hydroxycarbamide) is the cornerstone for children and adults with frequent crises or severe disease. It boosts production of fetal hemoglobin (HbF), which does not sickle and dilutes HbS in each red cell. The clinical goal is typically to achieve a fetal hemoglobin level of >20% (and ideally >30%), a threshold that drastically reduces HbS polymerization and prevents end-organ damage [17]. This reduces the frequency of pain crises and acute chest syndrome and improves survival. It is best established in HbSS and HbS/β⁰-thalassemia; its role in milder genotypes is guided by symptom burden. Treatment requires regular CBC monitoring because hydroxyurea can lower white cell counts.
Prophylactic penicillin is given to children with the severe genotypes, typically from around 2 months of age until at least age 5, to prevent pneumococcal sepsis. Patients also receive the standard childhood vaccines plus the pneumococcal (PCV13/PCV15 and PPSV23) and meningococcal vaccines.
What changed in 2023–2024
The treatment landscape has shifted significantly, and any current sickle cell resource needs to reflect this:
- Voxelotor (Oxbryta) is no longer available. Pfizer voluntarily withdrew it from all global markets on 25 September 2024 after clinical data showed an imbalance in vaso-occlusive crises and fatal events on the drug [1,2].
- Crizanlizumab (Adakveo) remains FDA-approved in the US, but its clinical use has sharply declined. The European Medicines Agency revoked its authorization in August 2023 after the STAND trial failed to confirm its benefit, leading many US centers to heavily restrict or abandon its use [3,11].
- L-glutamine (Endari), an oral powder thought to reduce oxidative stress in red cells, remains available and is approved for patients aged 5 and older.
- Mitapivat is an emerging oral pyruvate kinase (PK) activator. Following successful Phase 2/3 trials (such as the RISE UP study), it shows strong promise in improving hemoglobin levels and reducing pain crises by enhancing red blood cell energy metabolism and reducing HbS polymerization [12].
- Scheduled opioids, often via patient-controlled analgesia (PCA)
- NSAIDs for adjunct analgesia
- IV fluids for hydration
- Supplemental oxygen if needed
- Supplemental oxygen
- Broad-spectrum antibiotics
- Analgesia
- Exchange transfusion to reduce HbS below 30%
Guidance Shifts on Sildenafil and PAH Therapies
ASH 2025 guidance suggests that PAH-specific therapies, such as PDE-5 inhibitors (sildenafil, tadalafil) and soluble guanylate cyclase stimulators (riociguat), are safe in adults with SCD and confirmed pulmonary hypertension] [4]. This refines the older caution against using sildenafil in this population.
Pain management deserves special attention. Standard-of-care management now emphasizes multimodal analgesia to reduce reliance on high-dose opioids and mitigate the risk of opioid-induced hyperalgesia. This approach includes the early use of sub-dissociative ketamine infusions, targeted regional nerve blocks, and non-opioid adjuncts, alongside prompt, individualized opioid administration when appropriate [14].
Curative options
Bone marrow (stem cell) transplant can cure sickle cell disease. It is most successful when the donor is a matched sibling and the recipient is a child. The procedure carries real risks from the conditioning chemotherapy, graft-versus-host disease, and infection. Because it is intensive, it is generally reserved for more severe disease.
Gene therapies were FDA-approved in December 2023 for severe SCD:
- Casgevy (exagamglogene autotemcel) uses CRISPR-Cas9 to edit the patient's own blood stem cells, switching fetal hemoglobin production back on [10].
- Lyfgenia (lovotibeglogene autotemcel) uses a viral vector to add a working β-globin gene that produces an anti-sickling hemoglobin [9].
Both require conditioning chemotherapy and cost roughly USD 2–3 million per course. Access is therefore limited, and the global picture for sickle cell disease remains shaped less by what is technically possible than by who can reach it [5]. Furthermore, real-world rollout has been exceedingly slow. The entire process—from cell collection to manufacturing and conditioning—takes several months, and treatment centers face significant logistical and capacity limits [13].
Long-term monitoring
People with sickle cell disease need scheduled surveillance, not just reactive care. The intensity is tailored to genotype and symptom burden:
- TCD ultrasonography every year from age 2 to 16 in the severe genotypes to screen for stroke risk
- Screening brain MRI (without contrast) starting at school age to detect silent cerebral infarcts (SCIs). SCIs occur in up to 30% of children with HbSS and trigger the need for intensive disease-modifying therapy even if TCDs are normal [15]
- Annual eye exam to detect sickle retinopathy, especially important in HbSC disease
- Echocardiogram combined with NT-proBNP blood tests to screen for pulmonary hypertension. Elevated NT-proBNP and borderline echocardiogram findings prompt referral for a right heart catheterization, which is the diagnostic gold standard [16]
- Annual screening for microalbuminuria; if detected, ACE inhibitors or ARBs should be started immediately to preserve kidney function. Additionally, Cystatin C is increasingly preferred over standard serum creatinine to accurately track kidney function, as creatinine is notoriously unreliable in SCD due to hyperfiltration [16]
- Serum ferritin and, when available, cardiac and liver MRI T2* in patients on chronic transfusion, with iron chelation when iron overload develops
Pregnancy and sickle cell disease
Pregnancy raises the risk of vaso-occlusive crises, acute chest syndrome, preeclampsia, and preterm birth across all symptomatic genotypes. Care should be shared between hematology and obstetrics, with hydroxyurea typically stopped before conception and replaced by close monitoring and, in many cases, prophylactic transfusion programs.
When caregivers should seek emergency care
Take a person with sickle cell disease to the emergency department for any of the following:
- Fever above 38.5°C (101.3°F)
- Severe pain not relieved by home medications
- Chest pain, fast breathing, or shortness of breath
- Sudden weakness, slurred speech, or facial droop
- Sudden swelling and tenderness of the abdomen, especially with paleness (this can happen in older children and adults with HbSC, not just young children with HbSS)
- A painful erection lasting more than 2 hours
- Confusion or unusual sleepiness
Each of these can mark a treatable emergency: sepsis, acute chest syndrome, stroke, splenic sequestration, or priapism.
Frequently Asked Questions (FAQs)
What is the difference between sickle cell disease and sickle cell anemia?
Sickle cell disease (SCD) is the umbrella term for all inherited disorders in which red cells sickle because they carry hemoglobin S. Sickle cell anemia is the most common and most severe single form, caused by inheriting two sickle genes (HbSS). Other forms of SCD include HbSC disease and HbS/β-thalassemia, which share the same mechanism but often run a milder or different course. So all sickle cell anemia is sickle cell disease, but not all sickle cell disease is sickle cell anemia.
Is sickle cell disease the same as sickle cell trait?
No. Sickle cell disease means inheriting two abnormal β-globin genes and having a symptomatic condition. Sickle cell trait (HbAS) means inheriting one sickle gene and one normal gene. People with trait are usually healthy carriers and have measurable protection against severe malaria. Trait can rarely cause symptoms with extreme dehydration, high altitude, or intense exertion, but it is a carrier state, not a disease.
Can sickle cell disease be cured?
Yes, in selected patients. A matched stem cell transplant can cure the disease, especially when a sibling donor is available. Two FDA-approved gene therapies, Casgevy and Lyfgenia, became available in late 2023 for severe SCD. Both edit or supplement the patient's own stem cells. All three options require conditioning chemotherapy and carry real risks, and the gene therapies cost roughly USD 2–3 million per course, so access is currently the biggest barrier.
Why does sickle cell disease cause so much pain?
Sickled red cells are stiff and sticky. They jam in small blood vessels and cut off oxygen to nearby tissues. The starved tissues release inflammatory and pain signals. These episodes are called vaso-occlusive crises and are the leading reason people with SCD seek emergency care. They occur across all genotypes, though their frequency varies.
Is voxelotor (Oxbryta) still used for sickle cell disease?
No. Pfizer withdrew voxelotor worldwide on 25 September 2024 after data showed more vaso-occlusive crises and deaths in patients on the drug. The FDA advised clinicians to stop prescribing it and to switch patients to alternatives, most often hydroxyurea [1,2].
Why are people with sickle cell disease so prone to infection?
The spleen normally clears bacteria with a sugar capsule, such as Streptococcus pneumoniae. In severe SCD, repeated blockages scar the spleen so badly that it stops working by early childhood, a process called autosplenectomy. Without a working spleen, even minor infections can become life-threatening, which is why children with severe SCD take prophylactic penicillin and receive extra vaccines. In milder forms like HbSC, the spleen may keep working for longer.
Does sickle cell disease shorten life expectancy?
Yes, although the gap has narrowed and varies by genotype. People with severe forms have a shorter median life expectancy than the general population because of cumulative organ damage and infection risk; milder genotypes generally fare better. Outcomes are strongly tied to access to early diagnosis, hydroxyurea, transfusion programs, and comprehensive care [5].
Glossary of Medical Terms
- Sickle cell disease (SCD): the umbrella term for all inherited disorders in which red cells sickle due to hemoglobin S; includes HbSS, HbSC, and HbS/β-thalassemia.
- Sickle cell anemia: the most severe and common form of SCD, caused by inheriting two sickle genes (HbSS).
- Hemoglobinopathy: an inherited disorder of hemoglobin structure or production.
- Hemoglobin S (HbS): the abnormal hemoglobin underlying all forms of sickle cell disease.
- Hemoglobin C (HbC): another abnormal hemoglobin; when inherited with HbS it causes HbSC disease.
- Polymerization: the linking of HbS molecules into long fibers when oxygen is low.
- Vaso-occlusion: blockage of small blood vessels by stiff, sickled cells.
- Vaso-occlusive crisis (VOC): a sudden, severe pain episode from vaso-occlusion.
- Ischemia: lack of blood flow and oxygen to a tissue.
- Infarction: tissue death caused by prolonged ischemia.
- Chronic hemolysis: ongoing premature destruction of red blood cells.
- Reticulocyte: a young red blood cell; high counts indicate active marrow response.
- Autosplenectomy: loss of spleen function from repeated infarctions, typical of HbSS but less so of milder genotypes.
- Encapsulated bacteria: bacteria with an outer sugar coat (e.g., S. pneumoniae); cleared by a working spleen.
- Dactylitis (hand-foot syndrome): painful swelling of hands and feet, often the first SCD symptom in infants.
- Acute chest syndrome (ACS): life-threatening lung complication with new infiltrate, fever, and respiratory symptoms.
- Priapism: prolonged, painful erection unrelated to sexual stimulation; an emergency in SCD.
- Avascular necrosis (AVN): bone tissue death from interrupted blood supply; notably common in HbSC.
- Fetal hemoglobin (HbF): prenatal hemoglobin that does not sickle; raised by hydroxyurea.
- Hydroxyurea: oral medicine that raises HbF and reduces crises.
- Conditioning regimen: chemotherapy or radiation given before stem cell transplant or gene therapy.
- Chelation therapy: medication that removes excess iron, used in chronically transfused patients.
Disclaimer: This article is intended for educational and informational purposes only. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Pfizer Inc. (2024, September 25). Pfizer voluntarily withdraws all lots of sickle cell disease treatment OXBRYTA® (voxelotor) from worldwide markets.
- U.S. Food and Drug Administration. (2024, September 26). FDA alerting patients and health care professionals about the voluntary withdrawal of Oxbryta from the market due to safety concerns.
- European Medicines Agency. (2023, August). Revocation of authorisation for sickle cell disease medicine Adakveo.
- Liem, R. I., Verhovsek, M., Wun, T., & Lanzkron, S. (2025). ASH sickle cell disease CPKD guidelines report. Blood advances, 9(22), 5692–5694. https://doi.org/10.1182/bloodadvances.2025016989
- American Society of Hematology. (2024). ASH sickle cell disease research priorities — 2024 update.
- Kavanagh, P. L., Fasipe, T. A., & Wun, T. (2022). Sickle Cell Disease: A Review. JAMA, 328(1), 57–68. https://doi.org/10.1001/jama.2022.10233
- Ware, R. E., de Montalembert, M., Tshilolo, L., & Abboud, M. R. (2017). Sickle cell disease. Lancet (London, England), 390(10091), 311–323. https://doi.org/10.1016/S0140-6736(17)30193-9
- Williams, T. N., & Thein, S. L. (2018). Sickle Cell Anemia and Its Phenotypes. Annual review of genomics and human genetics, 19, 113–147. https://doi.org/10.1146/annurev-genom-083117-021320
- Kanter, J., Walters, M. C., Krishnamurti, L., Mapara, M. Y., Kwiatkowski, J. L., Rifkin-Zenenberg, S., Aygun, B., Kasow, K. A., Pierciey, F. J., Jr, Bonner, M., Miller, A., Zhang, X., Lynch, J., Kim, D., Ribeil, J. A., Asmal, M., Goyal, S., Thompson, A. A., & Tisdale, J. F. (2022). Biologic and Clinical Efficacy of LentiGlobin for Sickle Cell Disease. The New England journal of medicine, 386(7), 617–628. https://doi.org/10.1056/NEJMoa2117175
- Frangoul, H., Altshuler, D., Cappellini, M. D., Chen, Y. S., Domm, J., Eustace, B. K., Foell, J., de la Fuente, J., Grupp, S., Handgretinger, R., Ho, T. W., Kattamis, A., Kernytsky, A., Lekstrom-Himes, J., Li, A. M., Locatelli, F., Mapara, M. Y., de Montalembert, M., Rondelli, D., Sharma, A., … Corbacioglu, S. (2021). CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. The New England journal of medicine, 384(3), 252–260. https://doi.org/10.1056/NEJMoa2031054
- Rai, P., Desai, P. C., & Ataga, K. I. (2022). The Evolving Landscape of Drug Therapies for Sickle Cell Disease. Hematology/oncology clinics of North America, 36(6), 1285–1312. https://doi.org/10.1016/j.hoc.2022.06.008
- Idowu, M., Otieno, L., Dumitriu, B., Lobo, C. L. C., Thein, S. L., Andemariam, B., Nnodu, O. E., Inati, A., Glaros, A. K., Bartolucci, P., Colombatti, R., Taher, A. T., Abboud, M. R., Darbari, D., Ataga, K. I., Antmen, A. B., Kuo, K. H. M., de Souza Medina, S., Oluyadi, A., Iyer, V., … Smith, W. R. (2025). Safety and efficacy of mitapivat in sickle cell disease (RISE UP): results from the phase 2 portion of a global, double-blind, randomised, placebo-controlled trial. The Lancet. Haematology, 12(1), e35–e44. https://doi.org/10.1016/S2352-3026(24)00319-3
- Sharma, A., Kassim, A., Thompson, A., Williams, D. A., Liu, H. D., Boelens, J. J., LaBelle, J. L., Tisdale, J., de la Fuente, J., Kanter, J., Krishnamurti, L., Pecker, L. H., Walters, M. C., Eapen, M., Porteus, M., Bhatia, M., Soni, S., Corbacioglu, S., John, T. D., & Cancio, M. I. (2026). Gene Therapy for Sickle Cell Disease: Practice Recommendations from the American Society for Transplantation and Cellular Therapy and the International Society for Cell & Gene Therapy. Transplantation and cellular therapy, S2666-6367(26)00225-3. Advance online publication. https://doi.org/10.1016/j.jtct.2026.03.019
- Brandow, A. M., Carroll, C. P., Creary, S., Edwards-Elliott, R., Glassberg, J., Hurley, R. W., Kutlar, A., Seisa, M., Stinson, J., Strouse, J. J., Yusuf, F., Zempsky, W., & Lang, E. (2020). American Society of Hematology 2020 guidelines for sickle cell disease: management of acute and chronic pain. Blood advances, 4(12), 2656–2701. https://doi.org/10.1182/bloodadvances.2020001851
- DeBaun, M. R., Jordan, L. C., King, A. A., Schatz, J., Vichinsky, E., Fox, C. K., McKinstry, R. C., Telfer, P., Kraut, M. A., Daraz, L., Kirkham, F. J., & Murad, M. H. (2020). American Society of Hematology 2020 guidelines for sickle cell disease: prevention, diagnosis, and treatment of cerebrovascular disease in children and adults. Blood advances, 4(8), 1554–1588. https://doi.org/10.1182/bloodadvances.2019001142
- Liem, R. I., Lanzkron, S., D Coates, T., DeCastro, L., Desai, A. A., Ataga, K. I., Cohen, R. T., Haynes, J., Osunkwo, I., Lebensburger, J. D., Lash, J. P., Wun, T., Verhovsek, M., Ontala, E., Blaylark, R., Alahdab, F., Katabi, A., & Mustafa, R. A. (2019). American Society of Hematology 2019 guidelines for sickle cell disease: cardiopulmonary and kidney disease. Blood advances, 3(23), 3867–3897. https://doi.org/10.1182/bloodadvances.2019000916
- Yawn, B. P., Buchanan, G. R., Afenyi-Annan, A. N., Ballas, S. K., Hassell, K. L., James, A. H., Jordan, L., Lanzkron, S. M., Lottenberg, R., Savage, W. J., Tanabe, P. J., Ware, R. E., Murad, M. H., Goldsmith, J. C., Ortiz, E., Fulwood, R., Horton, A., & John-Sowah, J. (2014). Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA, 312(10), 1033–1048. https://doi.org/10.1001/jama.2014.10517
- Adesina, O. O., & Neumayr, L. D. (2019). Osteonecrosis in sickle cell disease: an update on risk factors, diagnosis, and management. Hematology. American Society of Hematology. Education Program, 2019(1), 351–358. https://doi.org/10.1182/hematology.2019000038