Key Takeaways
Aplastic anemia is a bone marrow failure disorder in which the marrow stops producing enough red blood cells, white blood cells, and platelets, leading to pancytopenia.
- Signs and symptoms ▾: Lethargy, pallor (anemia), infections especially mouth and throat (neutropenia), bruising, epistaxis, gum bleeding (thrombocytopenia)
- Pathophysiology of idiopathic aplastic anemia (IAA) ▾: Most cases are immune-mediated. Cytotoxic T cells attack hematopoietic stem cells, releasing interferon-gamma and TNF-alpha that suppress and destroy the marrow [3,6].
- Laboratory investigations and diagnosis ▾: Diagnosis starts with a complete blood count and a bone marrow biopsy is essential. Flow cytometry is used to check for a PNH clone, cytogenetic testing to rule out MDS, and chromosome breakage or telomere length tests in younger patients to rule out inherited bone marrow failure syndromes.
- Treatment and management ▾: First-line treatment for severe disease is either an allogeneic stem cell transplant from a matched sibling donor, or horse ATG combined with cyclosporine and eltrombopag for patients without a matched donor [1,2].
*Click ▾ for more information
What is aplastic anemia?
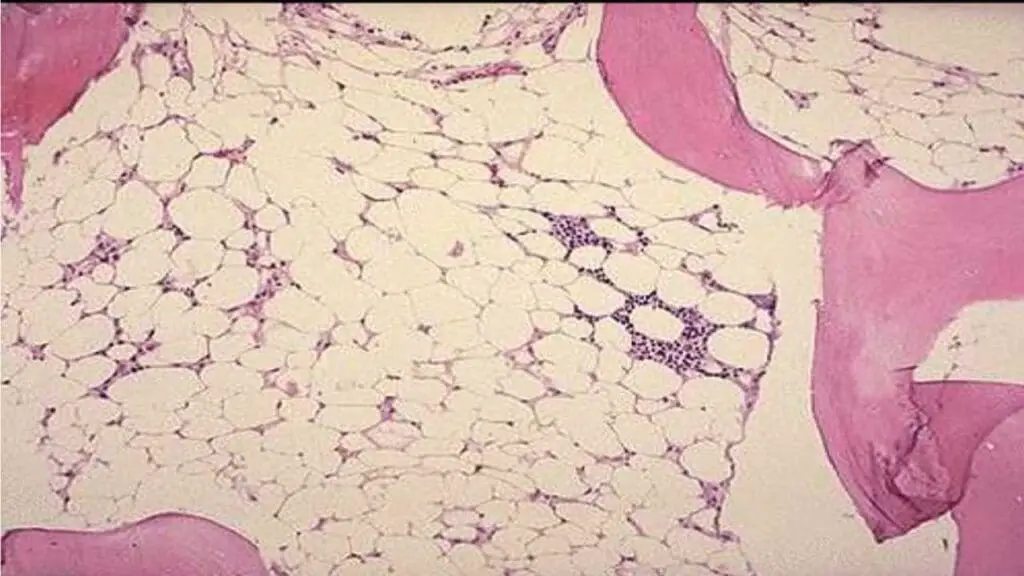
Aplastic anemia is a rare but serious blood disorder in which the bone marrow fails to produce enough red blood cells, white blood cells, and platelets [3]. This combined shortage is called pancytopenia (pan = all, cytopenia = low cell count).
The marrow does not just slow down. It empties out. On a biopsy, more than 75% of the marrow space is replaced by fat, with very few of the stem cells that should be there. The cells that remain are mostly lymphocytes and plasma cells, and they cannot make up for the loss [4].
This matters for patients because every symptom of aplastic anemia traces back to one of the three missing blood cell types. Low red cells (anemia) cause fatigue and pallor. Low white cells (leukopenia) cause infections. Low platelets (thrombocytopenia) cause bruising and bleeding.
What causes bone marrow failure?
Bone marrow failure is the broader umbrella term. Aplastic anemia is one cause, but not the only one. Other causes include marrow infiltration by cancer cells (myelophthisis), ineffective production of blood cells, and replacement of marrow by fibrous tissue as in myelofibrosis.
In aplastic anemia specifically, the problem is a major drop in the number of working hematopoietic stem cells. Either too few stem cells are present, or the remaining ones cannot divide properly because of an immune attack against them [3,6].
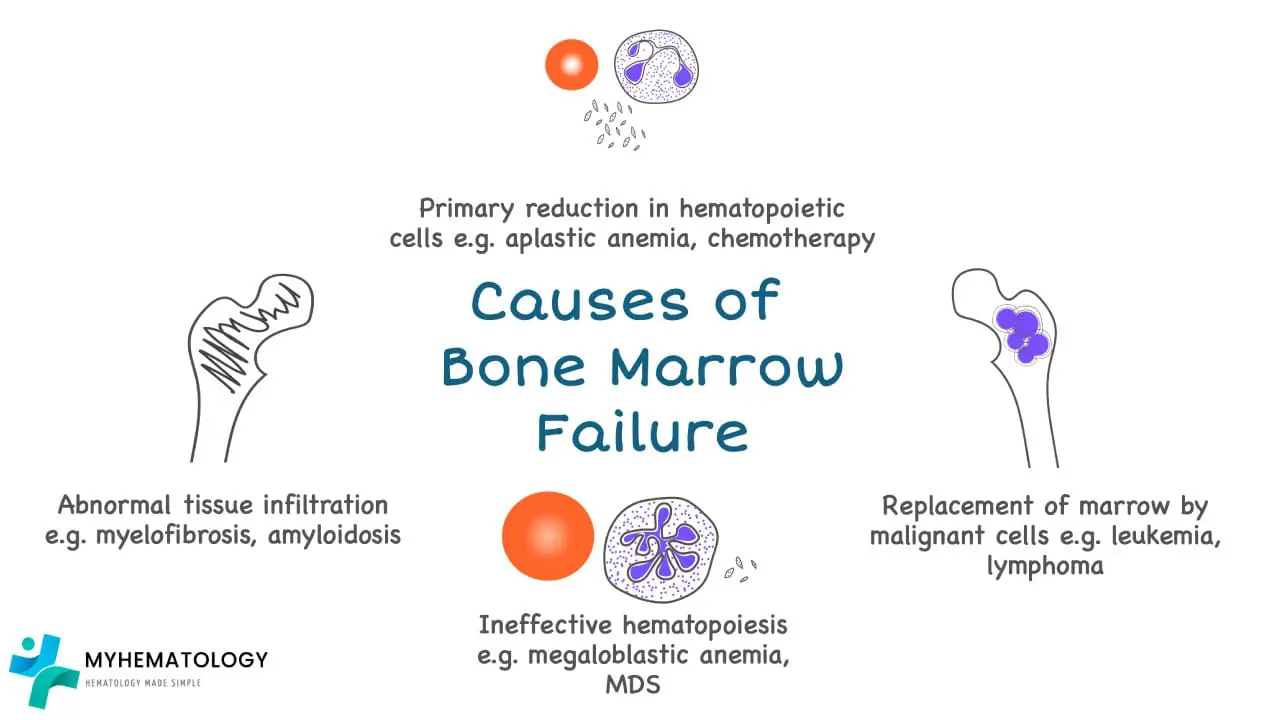
What are the causes of aplastic anemia?
Causes fall into two broad groups: acquired and inherited. When no cause can be identified — which happens often — the case is called idiopathic aplastic anemia.
Acquired Aplastic Anemia
This is by far the most common form, and most cases are autoimmune. The patient's own T cells mistakenly attack and destroy hematopoietic stem cells [3,6]. Known triggers include:
- Viral infections, including hepatitis viruses (the cause is usually unidentified rather than hepatitis B or C), parvovirus B19, and Epstein-Barr virus
- Drugs, classically chloramphenicol, phenytoin, carbamazepine, and some sulfonamides
- Chemicals and toxins, especially benzene and some pesticides
- Radiation and chemotherapy, which directly damage dividing marrow cells
- Pregnancy, which can trigger or worsen aplastic anemia in rare cases
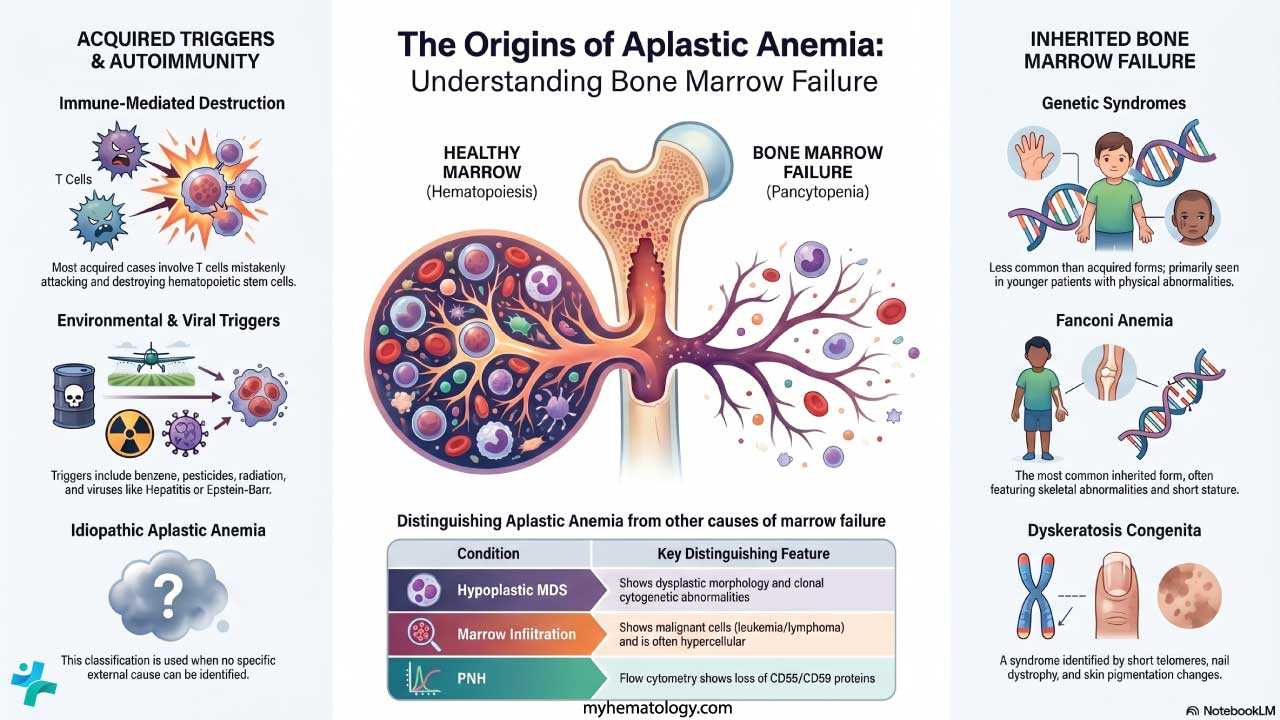
Inherited Aplastic Anemia
Inherited cases are much less common but are critical to recognize, especially in younger patients. They often present with extra physical features and have very different management. The key syndromes are:
- Fanconi anemia — the most common inherited form, often with skeletal abnormalities (especially of the thumbs), short stature, and skin pigment changes
- Dyskeratosis congenita — recognizable triad of nail dystrophy, abnormal skin pigmentation, and oral leukoplakia
- Shwachman-Diamond syndrome — pancreatic insufficiency, bone abnormalities, and neutropenia
These syndromes are screened for using chromosome breakage testing (for Fanconi anemia) and telomere length analysis (for dyskeratosis congenita) [4].
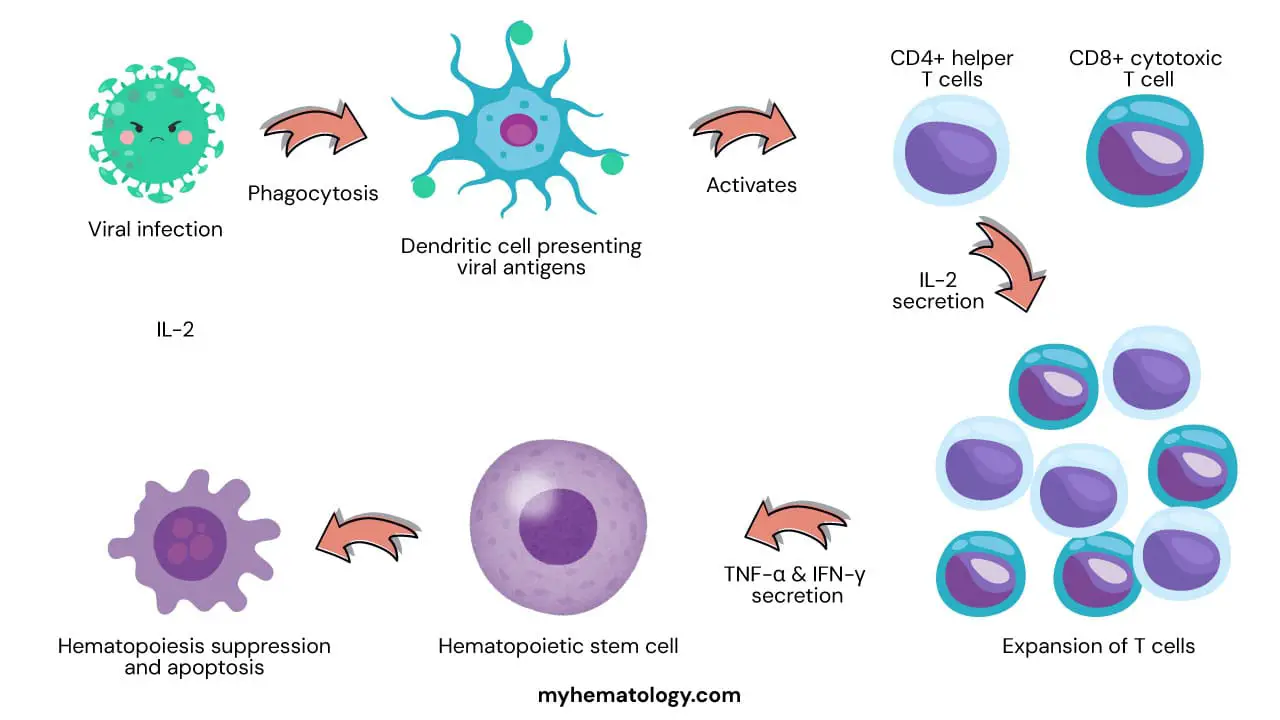
Pathophysiology
In idiopathic aplastic anemia, the leading model is autoimmune attack on hematopoietic stem cells. The sequence runs roughly like this [3,6]:
- A trigger such as a viral antigen, drug metabolite, or unknown self-antigen activates the immune system.
- Dendritic cells present this antigen to T cells in lymph nodes.
- CD4+ helper T cells release interleukin-2, which expands a population of CD8+ cytotoxic T cells.
- These activated T cells migrate to the bone marrow.
- Inside the marrow, they release interferon-gamma (IFN-γ) and tumor necrosis factor-alpha (TNF-α).
- These cytokines stop stem cells from dividing and push them into apoptosis (programmed cell death) by upregulating death receptors such as Fas.
- The stem cell pool collapses, and pancytopenia follows.
The same model explains why immunosuppressive therapy works: turn down the T cell response, and the surviving stem cells get a chance to recover.
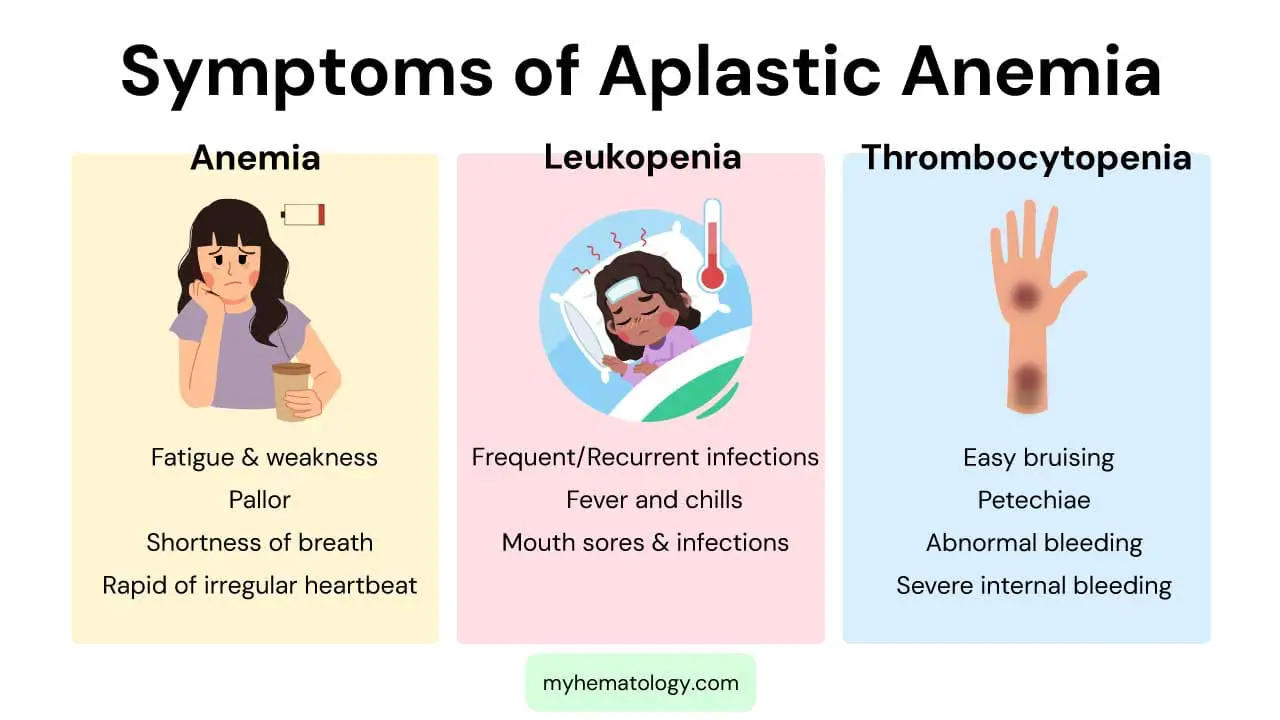
Signs and symptoms
Symptoms map directly onto which cell line is most affected. Severity depends on how low the counts have fallen.
From low red cells (anemia): fatigue, weakness, pallor, shortness of breath on exertion, rapid or irregular heartbeat.
From low white cells (leukopenia, especially neutropenia): frequent or unusually severe infections, unexplained fevers, mouth sores, and throat infections. Fever in a neutropenic patient is a medical emergency.
From low platelets (thrombocytopenia): easy bruising, petechiae (pinpoint red or purple spots on the skin), nosebleeds, gum bleeding, heavy menstrual bleeding, and rarely life-threatening internal bleeding.
For caregivers, the practical signs to watch for at home are new bruises, nosebleeds, fevers, and increasing tiredness — any of these warrant a same-day call to the medical team.
How is aplastic anemia diagnosed?
Diagnosis is one of exclusion. Many conditions cause pancytopenia, so the workup must rule them out [4].
Full blood count and peripheral blood film: Normochromic, normocytic, or slightly macrocytic anemia, low reticulocyte count, leukopenia, and thrombocytopenia. The blood film does not show abnormal or dysplastic cells.
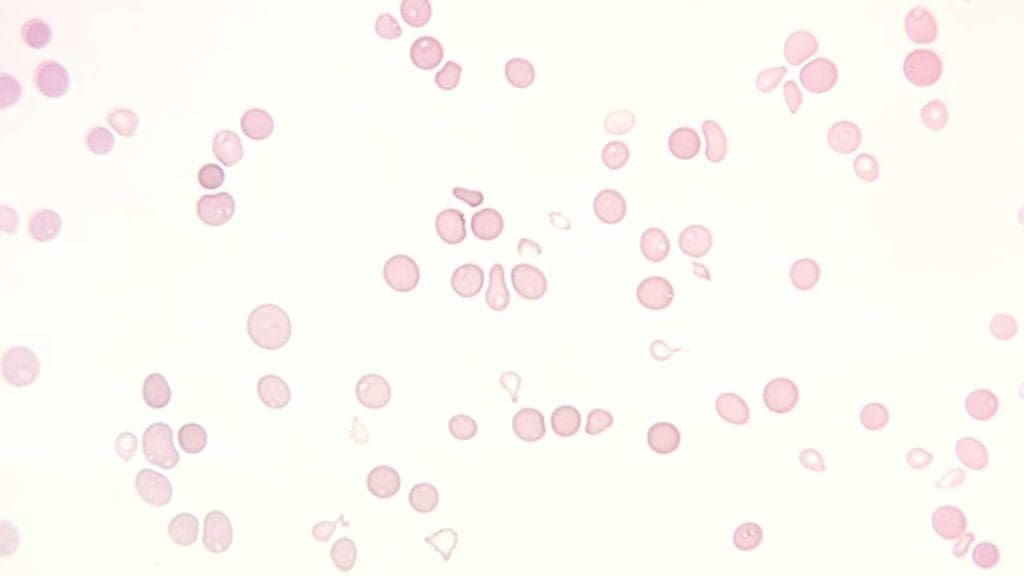
Bone marrow aspirate and trephine biopsy: The diagnostic core. The marrow is hypocellular (more than 75% fat), with the few remaining cells being lymphocytes and plasma cells. There are no dysplastic features and no abnormal infiltrates [4].
PNH flow cytometry: Every new aplastic anemia patient is screened for paroxysmal nocturnal hemoglobinuria using flow cytometry for GPI-anchored proteins (CD55, CD59) on red cells and granulocytes. Up to half of aplastic anemia patients carry a PNH clone [4].
Cytogenetic and molecular testing: Karyotyping and FISH look for chromosomal abnormalities that would suggest hypoplastic myelodysplastic syndrome (MDS). Next-generation sequencing (NGS) of myeloid genes is now a standard-of-care prognostic tool. Somatic mutations are found in about one-third of patients. Mutations in PIGA, BCOR, or BCORL1 predict a favorable response to immunosuppression. Conversely, mutations in ASXL1, DNMT3A, TP53, or RUNX1 carry a high risk of clonal evolution to MDS or AML, often prompting an earlier move to stem cell transplantation [10,15].
Inherited bone marrow failure screening: In patients under roughly 50 years old, chromosome breakage testing (Fanconi anemia) and telomere length assays (dyskeratosis congenita) are standard [4].
How is severity classified?
Once aplastic anemia is confirmed, severity is graded using the modified Camitta criteria [1,4]. This grading is what drives treatment.
Classification
- Marrow cellularity <25%
- Plus ≥2 of: neutrophils <0.5 × 10⁹/L, platelets <20 × 10⁹/L, reticulocytes <20 × 10⁹/L (or <60 × 10⁹/L automated)
- And neutrophils <0.2 × 10⁹/L
- Marrow cellularity <25%
- Plus ≥2 of: neutrophils <0.5 × 10⁹/L, platelets <20 × 10⁹/L, reticulocytes <20 × 10⁹/L (or <60 × 10⁹/L automated)
- Pancytopenia not meeting SAA criteria
Differential diagnosis
| Condition | Key Distinguishing Feature from AA |
|---|---|
| Hypoplastic MDS Clonal | Bone marrow shows dysplastic morphology and clonal cytogenetic abnormalities |
| PNH Flow Cytometry | Flow cytometry shows loss of CD55/CD59 on blood cells |
| Fanconi Anemia Inherited | Positive chromosome breakage test, often with physical anomalies |
| Dyskeratosis Congenita Inherited | Abnormally short telomeres, classic mucocutaneous triad |
| Large Granular Lymphocytic Leukemia Clonal | Clonal LGL population in blood and marrow |
| Marrow Infiltration Secondary | Marrow shows abnormal or malignant cells (leukemia, lymphoma, metastasis), often hypercellular |
Treatment and management
Modern aplastic anemia care is built around two questions: how severe is the disease, and is there a matched sibling donor? The answers determine the path [1,2].
Supportive care comes first
Every patient with aplastic anemia needs supportive care from day one. This is not optional as it keeps the patient alive while definitive treatment takes effect.
- Red cell and platelet transfusions to maintain hemoglobin above 7 g/dL and platelets above 10 × 10⁹/L. The platelet transfusion threshold is increased to 20 × 10⁹/L if the patient is febrile, or 50 × 10⁹/L if there is active bleeding [1,11]. All blood products should be leukoreduced and irradiated to prevent transfusion-associated graft-versus-host disease and to reduce alloimmunization, which is critical in transplant candidates.
- Antimicrobial prophylaxis with antibacterial, antifungal, and antiviral agents during deep neutropenia.
- Aggressive treatment of fever, which in a neutropenic patient is sepsis until proven otherwise.
- Iron chelation (for example, deferasirox) for patients on long-term transfusion programs to prevent iron overload. Not iron supplementation, which is not appropriate for aplastic anemia.
- Removal of any suspected causative drug or chemical at diagnosis.
First-line treatment: transplant or immunosuppression
For patients with severe or very severe aplastic anemia, first-line options are clear [1,2].
Allogeneic hematopoietic stem cell transplant (HSCT) from a matched sibling donor is preferred for patients under approximately 40 years old. In this group, 5-year survival exceeds 90% in modern series [1]. Furthermore, for pediatric and young adult patients (typically under 25–30 years old) who lack a matched sibling, a matched unrelated donor (MUD) transplant is increasingly recommended as a first-line option over immunosuppression. Modern high-resolution HLA-matching offers excellent cure rates while eliminating the risks of relapse or late clonal evolution [12].
Immunosuppressive therapy plus eltrombopag is first-line for patients without a matched sibling donor, or those who are older. The standard regimen is:
- Horse antithymocyte globulin (ATG) — depletes the autoreactive T cells driving the marrow attack. Horse ATG is preferred over rabbit ATG because of better response and survival [1].
- Cyclosporine — continues immune suppression for at least 6 months, often longer.
- Eltrombopag — a thrombopoietin receptor agonist that stimulates surviving stem cells to expand. Adding eltrombopag to ATG plus cyclosporine improves complete response rates and shortens time to recovery, as shown in the RACE trial [2,5,7]. For patients who experience eltrombopag-induced liver toxicity or do not respond, alternative TPO-RAs such as romiplostim (a weekly subcutaneous injection) are highly effective and are now routinely utilized in both upfront and refractory settings [13].
This three-drug combination became standard after 2022 and is now the first-line immunosuppressive backbone in major guidelines [1].
Second-line and refractory disease
Patients who do not respond to first-line immunosuppression may be offered:
- Allogeneic HSCT from a matched unrelated donor or a haploidentical (half-matched family) donor. Haploidentical transplants have been revolutionized by post-transplant cyclophosphamide (PTCy) protocols, which reliably prevent severe graft-versus-host disease (GVHD). Due to this excellent safety profile, haplo-transplants are now highly successful and are frequently preferred over a second round of immunosuppression [9,14].
- Repeat course of immunosuppression with continued eltrombopag.
Long-term monitoring and clonal evolution
Even after recovery, patients with aplastic anemia need long-term follow-up. A small but important subset develops clonal evolution to myelodysplastic syndrome, acute myeloid leukemia, or expansion of a PNH clone [7]. Regular blood counts, periodic flow cytometry, and repeat marrow assessment when indicated are part of survivorship care.
Prognosis
Outcomes have improved sharply over the past two decades. Young patients who receive a matched sibling donor transplant now have 5-year survival above 90% [1]. Patients on horse ATG, cyclosporine, and eltrombopag achieve overall response rates of around 70% and 5-year survival in the 70–80% range [2,7]. Older age, very severe disease, infection at diagnosis, and lack of response to first-line therapy all reduce survival.
A note for patients and caregivers
Aplastic anemia is rare, the diagnosis can be frightening, and treatment is long. The good news is that modern care — transplant when feasible, eltrombopag-based immunosuppression when not — has changed the outlook substantially. Most young patients can expect to recover blood counts and live a normal lifespan, though follow-up continues for years afterward.
Frequently Asked Questions (FAQs)
What is aplastic anemia in simple terms?
Aplastic anemia is a rare bone marrow failure disorder. The bone marrow stops making enough red blood cells, white blood cells, and platelets. This leads to fatigue, frequent infections, and easy bleeding. Most cases are caused by the immune system mistakenly attacking the bone marrow, but some are inherited or triggered by drugs, toxins, viruses, or radiation.
How is aplastic anemia diagnosed?
Diagnosis starts with a complete blood count showing low red cells, white cells, and platelets, and a low reticulocyte count. A bone marrow biopsy is essential and shows a hypocellular (mostly fat) marrow with no abnormal cells. Doctors also run flow cytometry to check for a PNH clone, cytogenetic testing to rule out MDS, and chromosome breakage or telomere length tests in younger patients to rule out inherited bone marrow failure syndromes such as Fanconi anemia or dyskeratosis congenita.
What is the first-line treatment for severe aplastic anemia?
Treatment depends on age and donor availability. Younger patients with a matched sibling donor are usually offered an allogeneic stem cell transplant first. Patients without a sibling donor, or those who are older, receive immunosuppressive therapy. The current standard regimen is horse antithymocyte globulin (ATG) combined with cyclosporine and eltrombopag, a thrombopoietin receptor agonist. This combination produces faster and more complete recovery of blood counts than the older two-drug regimen.
Can aplastic anemia turn into leukemia?
Aplastic anemia does not transform directly into leukemia. However, a small proportion of patients, particularly those treated long-term with immunosuppression, develop a related condition called clonal evolution. This can progress to myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML). Regular blood monitoring after recovery is important to detect this early.
What is the survival rate for aplastic anemia?
Outcomes have improved substantially. Young patients who undergo a matched sibling donor stem cell transplant have 5-year survival rates above 90%. Patients treated with modern immunosuppression plus eltrombopag have 5-year survival rates of roughly 70–80%. Older age, very severe disease, infection at diagnosis, and lack of treatment response all lower survival.
How can a patient with aplastic anemia stay safe at home?
Caregivers and patients should focus on infection and bleeding prevention. Practice strict hand hygiene, avoid sick contacts, avoid raw or undercooked foods, and report any fever above 38°C immediately. Use a soft toothbrush, avoid contact sports, and keep a current list of medications. Follow transfusion appointments closely and report new bruising, nosebleeds, or unusual fatigue to the medical team.
Glossary of Related Medical Terms
- Aplasia: Failure of an organ or tissue to develop or function. In aplastic anemia, the bone marrow fails to make blood cells.
- Bone marrow: The spongy tissue inside large bones where blood cells are made.
- Clonal evolution: The development, over time, of an abnormal cell line that can lead to a different blood disorder such as MDS, AML, or PNH.
- Cytopenia: A reduced number of one or more types of blood cells.
- Erythropoiesis: The body's process of making red blood cells.
- Hematopoietic stem cells (HSCs): Master blood-forming cells in the bone marrow that give rise to all red cells, white cells, and platelets.
- Immunosuppressive therapy (IST): Medication that calms the immune system. In AA, IST is used because the immune system is attacking the bone marrow.
- Modified Camitta criteria: The standard scoring system used to classify AA as non-severe, severe, or very severe.
- Neutropenia: A low neutrophil count, which raises infection risk.
- Pancytopenia: Low counts of all three blood cell lines: red cells, white cells, and platelets.
- Paroxysmal nocturnal hemoglobinuria (PNH): A related blood disorder that can co-exist with AA; screened for using flow cytometry.
- Reticulocytes: Young red blood cells. A low reticulocyte count signals the marrow is not making new red cells.
- Thrombocytopenia: A low platelet count, which causes easy bruising and bleeding.
- Thrombopoietin receptor agonist: A drug class (e.g., eltrombopag) that mimics the natural hormone thrombopoietin to stimulate blood cell production.
Disclaimer: This article is intended for educational and informational purposes only . It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Kulasekararaj A, Cavenagh J, Dokal I, Foukaneli T, Gandhi S, Garg M, et al. Guidelines for the diagnosis and management of adult aplastic anaemia: A British Society for Haematology Guideline. Br J Haematol. 2024;204(3):784–804. https://doi.org/10.1111/bjh.19236
- Peffault de Latour, R., Kulasekararaj, A., Iacobelli, S., Terwel, S. R., Cook, R., Griffin, M., Halkes, C. J. M., Recher, C., Barraco, F., Forcade, E., Vallejo, J. C., Drexler, B., Mear, J. B., Smith, A. E., Angelucci, E., Raymakers, R. A. P., de Groot, M. R., Daguindau, E., Nur, E., Barcellini, W., … Severe Aplastic Anemia Working Party of the European Society for Blood and Marrow Transplantation (2022). Eltrombopag Added to Immunosuppression in Severe Aplastic Anemia. The New England journal of medicine, 386(1), 11–23. https://doi.org/10.1056/NEJMoa2109965
- Young N. S. (2018). Aplastic Anemia. The New England journal of medicine, 379(17), 1643–1656. https://doi.org/10.1056/NEJMra1413485
- DeZern, A. E., & Churpek, J. E. (2021). Approach to the diagnosis of aplastic anemia. Blood Advances, 5(12), 2660–2671. https://doi.org/10.1182/bloodadvances.2021004345
- Townsley, D. M., Scheinberg, P., Winkler, T., Desmond, R., Dumitriu, B., Rios, O., Weinstein, B., Valdez, J., Lotter, J., Feng, X., Desierto, M., Leuva, H., Bevans, M., Wu, C., Larochelle, A., Calvo, K. R., Dunbar, C. E., & Young, N. S. (2017). Eltrombopag Added to Standard Immunosuppression for Aplastic Anemia. The New England journal of medicine, 376(16), 1540–1550. https://doi.org/10.1056/NEJMoa1613878
- Schoettler, M. L., & Nathan, D. G. (2018). The Pathophysiology of Acquired Aplastic Anemia: Current Concepts Revisited. Hematology/oncology clinics of North America, 32(4), 581–594. https://doi.org/10.1016/j.hoc.2018.03.001
- Patel, B. A., Groarke, E. M., Lotter, J., Shalhoub, R., Gutierrez-Rodrigues, F., Rios, O., Quinones Raffo, D., Wu, C. O., & Young, N. S. (2022). Long-term outcomes in patients with severe aplastic anemia treated with immunosuppression and eltrombopag: a phase 2 study. Blood, 139(1), 34–43. https://doi.org/10.1182/blood.2021012130
- Bacigalupo A. (2017). How I treat acquired aplastic anemia. Blood, 129(11), 1428–1436. https://doi.org/10.1182/blood-2016-08-693481
- DeZern, A. E., Eapen, M., Wu, J., Talano, J. A., Solh, M., Dávila Saldaña, B. J., Karanes, C., Horwitz, M. E., Mallhi, K., Arai, S., Farhadfar, N., Hexner, E., Westervelt, P., Antin, J. H., Deeg, H. J., Leifer, E., Brodsky, R. A., Logan, B. R., Horowitz, M. M., Jones, R. J., … Pulsipher, M. A. (2022). Haploidentical bone marrow transplantation in patients with relapsed or refractory severe aplastic anaemia in the USA (BMT CTN 1502): a multicentre, single-arm, phase 2 trial. The Lancet. Haematology, 9(9), e660–e669. https://doi.org/10.1016/S2352-3026(22)00206-X
- Babushok D. V. (2018). A brief, but comprehensive, guide to clonal evolution in aplastic anemia. Hematology. American Society of Hematology. Education Program, 2018(1), 457–466. https://doi.org/10.1182/asheducation-2018.1.457
- Kaufman, R. M., Djulbegovic, B., Gernsheimer, T., Kleinman, S., Tinmouth, A. T., Capocelli, K. E., Cipolle, M. D., Cohn, C. S., Fung, M. K., Grossman, B. J., Mintz, P. D., O'Malley, B. A., Sesok-Pizzini, D. A., Shander, A., Stack, G. E., Webert, K. E., Weinstein, R., Welch, B. G., Whitman, G. J., Wong, E. C., … AABB (2015). Platelet transfusion: a clinical practice guideline from the AABB. Annals of internal medicine, 162(3), 205–213. https://doi.org/10.7326/M14-1589
- Scheinberg, P., & Kulasekararaj, A. G. (2024). Consensus recommendations for severe aplastic anemia. Blood advances, 8(21), 5719–5720. https://doi.org/10.1182/bloodadvances.2024013529
- Jang, J. H., Tomiyama, Y., Miyazaki, K., Nagafuji, K., Usuki, K., Uoshima, N., Fujisaki, T., Kosugi, H., Matsumura, I., Sasaki, K., Kizaki, M., Sawa, M., Hidaka, M., Kobayashi, N., Ichikawa, S., Yonemura, Y., Enokitani, K., Matsuda, A., Ozawa, K., Mitani, K., … Nakao, S. (2021). Efficacy and safety of romiplostim in refractory aplastic anaemia: a Phase II/III, multicentre, open-label study. British journal of haematology, 192(1), 190–199. https://doi.org/10.1111/bjh.17190
- DeZern, A. E., & Brodsky, R. A. (2018). Haploidentical Donor Bone Marrow Transplantation for Severe Aplastic Anemia. Hematology/oncology clinics of North America, 32(4), 629–642. https://doi.org/10.1016/j.hoc.2018.04.001
- Wood, H. J., & Marsh, J. C. W. (2019). Somatic mutations in aplastic anemia: Significance for classification, therapy, and outcome. HemaSphere, 3(Suppl), 10-12. https://doi.org/10.1097/HS9.0000000000000212