Key Takeaways
Primary hemostasis is the body’s rapid response to a broken blood vessel in the physiology of hemostasis, forming a platelet plug through platelet adhesion and platelet aggregation before the secondary hemostasis (coagulation cascade) kicks in.
- Key Players ▾:
- Platelets: They adhere to the injured vessel wall, activate, and aggregate to form a platelet plug in primary hemostasis.
- Von Willebrand factor (vWF): A protein bridge, linking platelets to the exposed collagen in the injured vessel wall.
- Coagulation factors: Supporting players, further stabilizing the clot through fibrin formation.
- The Steps in Primary Hemostasis ▾:
- Platelet Adhesion: Platelets stick to vWF and exposed collagen like glue.
- Platelet Activation: Platelets change shape, sprout spiky pseudopods, and release signals to recruit more platelets.
- Platelet Aggregation: Platelets grab onto each other through fibrinogen bridges, forming a dense mesh.
- Clot Retraction: Platelets contract, pulling the wound edges together and strengthening the plug.
- Disorders of Primary Hemostasis ▾:
- Quantitative Abnormalities
- Thrombocytopenia: Common causes include aplastic anemia, immune thrombocytopenia, and bone marrow infiltration.
- Qualitative Abnormalities
- Platelet dysfunction: Examples include von Willebrand disease, Bernard-Soulier syndrome, and Glanzmann thrombasthenia.
- Vascular Wall Abnormalities
- Defects in the endothelium or subendothelial structures affecting vWF binding and platelet adhesion. Examples include endothelial dysfunction, vascular injury, and congenital disorders like Ehlers-Danlos syndrome.
- Quantitative Abnormalities
- General Investigations for Primary Hemostasis ▾:
- Complete Blood Count (CBC): Checks platelet count, a key indicator of bleeding risk.
- Peripheral Blood Smear: Visualizes platelet morphology for clues about potential disorders.
- Bleeding Time Test: Assesses platelet adhesion and aggregation at the injury site.
- Specific Assays: Measure vWF levels and function, platelet aggregation patterns, or test for genetic mutations in specific disorders.
- Treatment and Management Strategies ▾:
- Address the underlying cause: Treat infections, correct deficiencies, or manage autoimmune conditions.
- Platelet transfusion: For severe thrombocytopenia with active bleeding.
- Antifibrinolytic drugs: Stabilize existing clots by preventing fibrin breakdown.
- Desmopressin: Promote vWF release in mild vWF deficiency.
- Bone marrow transplantation: Potential cure for severe inherited platelet disorders.
*Click ▾ for more information
Introduction
Hemostasis is the coordinated physiological mechanisms that prevent and stop blood loss after injury. This coordinated response involves two main phases:
1. Primary Hemostasis: Tiny platelets rush to the scene, sticking to the injured vessel wall and each other (platelet aggregation), forming a temporary plug at the injury site within seconds to stop the bleeding.
2. Secondary Hemostasis: The slow and steady reinforcement team, involving the coagulation cascade, forming a tougher, fibrin-based clot to stabilize the plug and promote healing.
Platelets in Primary Hemostasis
Platelets, the unsung heroes of primary hemostasis, are more than just miniature blood cells. They’re anucleated cell fragments, remnants of megakaryocytes, the giant cells of the bone marrow. Though lacking a nucleus, they pack a powerful punch in their tiny bodies, orchestrating the initial, rapid response to injury.
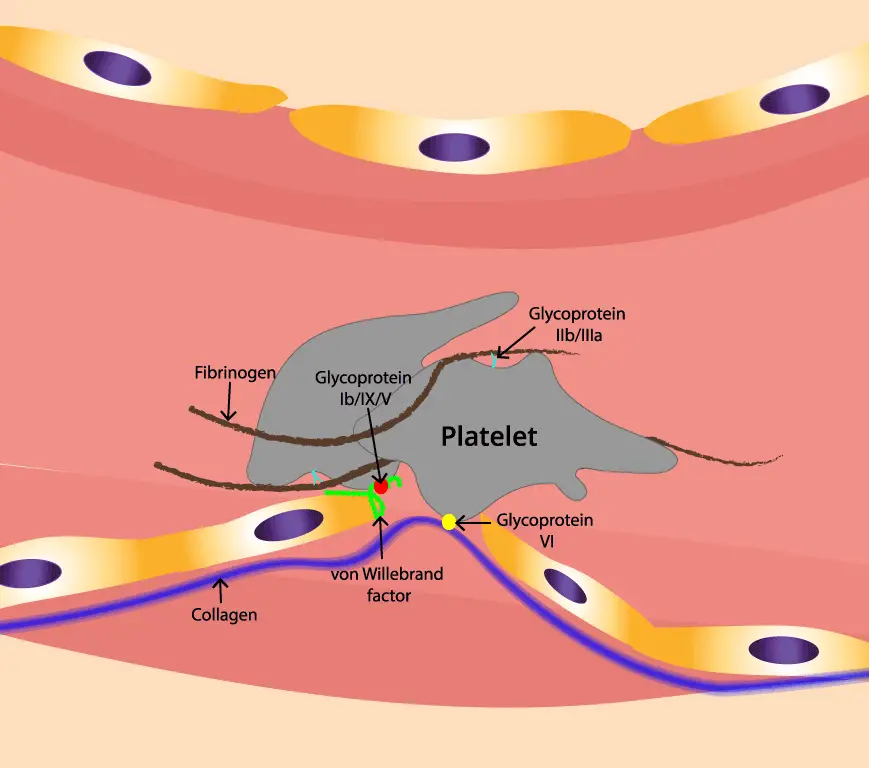
Platelet Adhesion: Platelets patrol the blood vessels for exposed collagen of an injured vessel. They possess specific surface receptors that bind tightly to collagen with the help of a bridge-building protein called von Willebrand factor. This initial adhesion acts as an anchor, slowing down the blood flow and attracting more platelets to the scene.
Platelet Activation: Once anchored, platelets undergo a dramatic transformation. They change shape, sprouting spiky extensions to grab onto other platelets and the vessel wall. They release a potent cocktail of chemicals from their internal granules, attracting more platelets and triggering the next crucial step: aggregation.
Platelet Aggregation: Picture a domino effect. Each activated platelet acts as a glue trap, grabbing onto nearby platelets via fibrinogen. This chain reaction builds a dense platelet plug, like a living dam, effectively blocking the leak and preventing further blood loss.
Mechanism of Primary Hemostasis
Vascular Spasm
Before platelets even arrive in the event of an injury, the blood vessel walls violently contract at the site of injury. This rapid vasoconstriction is nature’s first line of defense in primary hemostasis, buying precious time for the platelet plug to form.

Key Players
- Endothelin: This potent vasoconstrictor peptide is produced by the injured endothelial cells themselves. Think of it as the alarm bell, sending a distress signal to the surrounding muscle cells. Endothelin activates specific receptors on the smooth muscle cells in the vessel wall, triggering two main pathways:
- The Gq/11-Protein Kinase C Pathway: This pathway leads to increased calcium levels in the smooth muscle cells. Calcium activates enzymes called kinases, which add phosphate groups to proteins within the muscle cells. This phosphorylation activates the contractile machinery within the muscle cells, causing them to contract and squeeze the vessel.
- The Rho-Kinase Pathway: This pathway promotes the formation of actin fibers, the muscles’ contractile machinery, further tightening the vessel. Phosphorylated proteins, like myosin and actin, form intricate cross-bridges within the muscle cells. These cross-bridges pull and slide against each other, squeezing the vessel wall and constricting its lumen.
- Neural Reflexes: Sensory nerves near the injury send distress signals to the brainstem, which activates the sympathetic nervous system. This system releases neurotransmitters like norepinephrine that binds to smooth muscle receptors. Norepinephrine binds to alpha-adrenergic receptors on smooth muscle cells, activating the Gq/11 pathway and potentiating the calcium-induced contraction, reinforcing the vasoconstriction.
The combined effects of endothelin and neural reflexes lead to a rapid and forceful vasoconstriction at the injury site, essential for primary hemostasis. This reduces blood flow to the damaged area, minimizing the amount of blood lost and creating a calmer environment for the platelets to do their job.
Platelet Adhesion in Primary Hemostasis
When a blood vessel is injured, the smooth endothelial lining tears away, revealing the underlying collagen fibrils. These fibrils, like rough ropes, act as a sticky carpet for platelets to grab onto in primary hemostasis.
Then comes the von Willebrand factor (vWF). vWF is a large, multi-domain protein produced by endothelial cells and megakaryocytes. vWF exists in different forms in the blood. However, in the face of injury, vWF undergoes a crucial activation process.
Activation in High-Flow Zones
Flowing blood creates shear stress, and in areas with high flow, this stress stretches the vWF molecule, unfolding its A1 domain. This unfolding exposes a high-affinity binding site for the glycoprotein Ibα (GPIbα) receptor on the platelet surface.
This stretching exposes hidden binding sites within vWF, allowing a specific enzyme called ADAMTS13 to cleave the molecule. This cleavage releases smaller, more active vWF fragments that are ready to bind. These active fragments then bind to each other, forming long, thread-like structures called multimers.
The Collagen Catch
vWF also possesses distinct binding sites for exposed collagen fibrils. These sites, particularly in the A3 domain, interact with specific collagen components like glycoprotein (Gp) VI on the platelet surface.
The Two-Step Handshake in Primary Hemostasis
- Step 1 (Initial Tethering): The stretched A1 domain on vWF grabs onto GPIbα on a passing platelet, temporarily tethering it to the vessel wall. This initial contact slows down the platelet and allows further interactions.
- Step 2 (Firm Adhesion): The exposed collagen sites on vWF engage with GpVI receptors on the platelet, leading to a stronger, more definitive adhesion. This activates the platelet, setting the stage for the next steps in primary hemostasis.
Platelet Activation in Primary Hemostasis
As a tiny platelet encounters the scene of a blood vessel injury, it undergoes a dramatic metamorphosis, morphing from a passive bystander into a powerful actor in the primary hemostasis play.
Platelets have two main types of granules
- Dense granules: These store small molecules like adenosine diphosphate (ADP), serotonin, and calcium, crucial for platelet aggregation and vasoconstriction.
- ADP: Activates nearby platelets, triggering further aggregation and amplifying the response in primary hemostasis.
- Serotonin: Contributes to vasoconstriction, reducing blood flow to the injured area.
- Calcium: Drives platelet activation and granule release.
- Alpha granules: These hold a diverse array of proteins with various functions, including:
- P-selectin: Adheres to white blood cells, initiating inflammatory responses and wound healing.
- Fibrinogen: Bridges between activated platelets, forming the core of the platelet plug in primary hemostasis.
- vWF: Helps anchor platelets to collagen and recruit other platelets to the growing plug.
- Platelet Factor 4 (PF-4): This antibacterial peptide (ABP), accounting for a significant portion of platelet granule contents, exhibits broad-spectrum activity against bacteria, fungi, and even viruses. PF-4 disrupts microbial membranes, essentially poking holes in their armor, leading to rapid cell death.
- Cathelicidin LL-37: This potent ABP targets bacterial membranes similarly to PF-4 but also neutralizes bacterial toxins, attracts immune cells to the site of infection, and promotes wound healing.
- Human Beta-Defensin 3 (hBD-3): This relatively small ABP works by disrupting bacterial cell walls and inhibiting their growth. Interestingly, hBD-3 also exhibits anti-inflammatory properties, helping minimize tissue damage during the fight against invaders.
- Thrombocidin-1 (TC-1): This unique ABP targets bacteria hiding within fibrin clots, a common bacterial refuge during platelet aggregation. TC-1 punches holes in the fibrin mesh, flushing out pathogens and exposing them to other ABPs and immune cells.
- Platelet-Derived Growth Factor (PDGF): This peptide comes in several isoforms (AA, BB, AB), each targeting specific cell types. PDGFs stimulate vascular smooth muscle cells, endothelial cells, and fibroblasts, promoting their growth and migration. This contributes to:
- Angiogenesis: The formation of new blood vessels, essential for delivering oxygen and nutrients to the repair site.
- Fibrosis: Scar tissue formation, providing structural support and wound closure.
- Transforming Growth Factor-β (TGF-β): This versatile growth factor plays a dual role in wound healing. Initially, it promotes inflammation and attracts immune cells to fight infection. Later, it switches gears, stimulating extracellular matrix production and collagen deposition, contributing to scar tissue formation and wound closure.
- Vascular Endothelial Growth Factor (VEGF): This growth factor, as its name suggests, specifically targets endothelial cells, promoting their proliferation and migration. This leads to:
- Angiogenesis: Similar to PDGF, VEGF plays a crucial role in forming new blood vessels to support the healing process.
- Vascular permeability: VEGF can increase the permeability of existing blood vessels, allowing immune cells and nutrients to reach the injured site more efficiently.
- Epidermal Growth Factor (EGF): This growth factor targets keratinocytes, the main cell type in the epidermis (outermost skin layer). EGF stimulates their proliferation and migration, promoting:
- Re-epithelialization: Growth of new skin cells to cover the wound and restore the skin barrier.
- Wound contraction: EGF also plays a role in wound contraction, helping the wound edges pull together and close the gap.
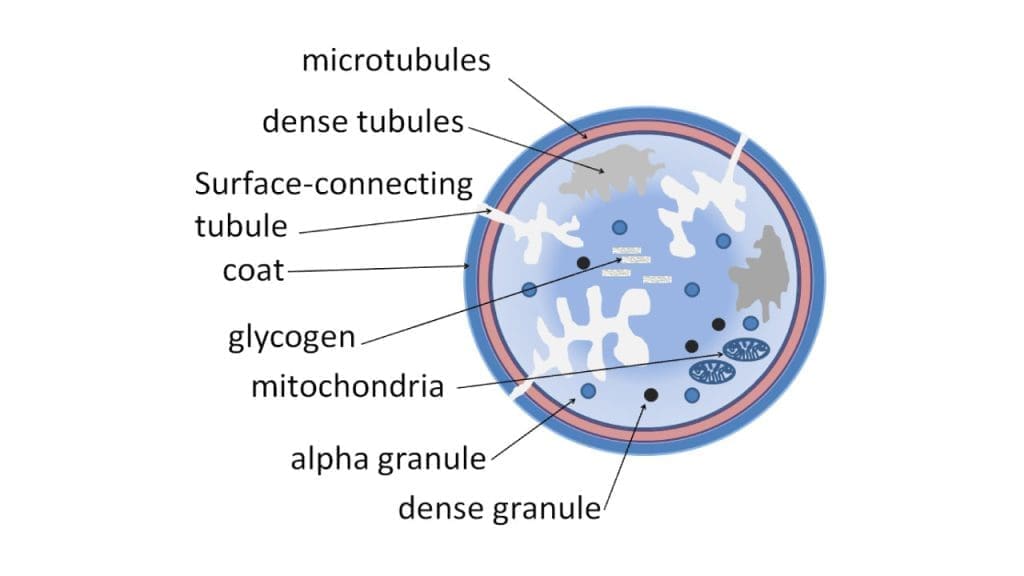
The Triggers
Binding to collagen and vWF triggers the release of ADP (adenosine diphosphate) from the platelet’s dense granules for use in primary hemostasis. ADP acts as a signal, binding to specific receptors on the platelet surface.
The Conformational Change
- Receptor engagement: Specific receptors on the platelet surface, like GPIb and GPIIb/IIIa, bind to collagen, vWF, or other activated platelets.
- Calcium influx: Channels open, allowing a surge of calcium ions into the platelet. Calcium acts as a second messenger, amplifying the activation signal.
- Phosphorylation: Enzymes called kinases go on a phosphorylation spree, modifying proteins within the platelet and altering their functions.
- Shape shift: The platelet cytoskeleton rearranges, leading to the iconic spiky morphology, allowing for better interaction with other platelets and the vessel wall.
- Granule release: The internal membranes of the granules fuse with the platelet plasma membrane, releasing their contents into the surrounding environment.
Platelet Aggregation in Primary Hemostasis
After the platelet activation in primary hemostasis, fibrinogen takes center stage. This large, multi-domain protein acts like a bridge, connecting activated platelets to each other. It has two key binding sites:
- Fibrinogen Binding Sites on Platelets: Located on the platelet surface, these sites bind specifically to fibrinogen’s Aα and Bβ chains, forming a secure connection in platelet aggregation.
- Knob-and-Hole Mechanism: Think of fibrinogen as a Y-shaped molecule. The Aα and Bβ chains are the “knobs,” while the central C-domain is the “hole.” Activated platelets possess complementary “hole-like” receptors that fit perfectly onto these knobs, further strengthening the bond of platelet aggregation.
The Platelet Aggregation Cascade in Primary Hemostasis
- Initial Tethering: The released fibrinogen molecules act like sticky strands, tethering nearby activated platelets to each other through their fibrinogen binding sites. This initial connection in platelet aggregation, though weak, slows down the platelets and allows for further interactions in primary hemostasis.
- Amplification Through ADP: ADP released from activated platelets acts like a siren call, attracting more circulating platelets to the growing platelet aggregation. These attracted platelets bind to fibrinogen bridges already formed, further strengthening and expanding the platelet plug in primary hemostasis.
- Positive Feedback Loop: As more platelets join the party, they themselves become activated by the surrounding ADP and release fibrinogen. This creates a positive feedback loop, leading to a rapid and exponential increase in platelet aggregation in the platelet plug formation.
- Final Stabilization: As the platelet plug grows, other adhesive molecules like von Willebrand factor and P-selectin come into play, providing additional stability and anchoring the platelet plug to the injured vessel wall in primary hemostasis.
The Outcome
This cascade of activation, recruitment, and binding culminates in a dense and sturdy platelet plug. This platelet plug in primary hemostasis acts as a temporary dam, effectively sealing the leak and preventing further blood loss until the secondary phase of hemostasis takes over with fibrin clot formation.
Clot Retraction in Primary Hemostasis
Clot retraction is the final stage of primary hemostasis, where the newly formed platelet plug contracts, pulling the wound edges closer together and strengthening the clot. This process is vital for effective wound healing and preventing excessive blood loss.
Key Players and Mechanism
- Myosin and Actin: These contractile proteins within platelets, similar to those found in muscle cells, are responsible for clot retraction.
- Calcium: The release of calcium from platelet granules activates myosin, enabling it to bind to actin filaments and initiate the contraction process.
- Thrombin: The enzyme thrombin, generated during secondary hemostasis, plays a crucial role in stabilizing the fibrin clot and enhancing clot retraction.
Steps of Clot Retraction
- Myosin Activation: Calcium ions bind to calmodulin, a regulatory protein. The calcium-calmodulin complex activates myosin, enabling it to bind to actin filaments.
- Cross-bridge Formation: Myosin heads bind to actin filaments, forming cross-bridges.
- Contraction: Myosin heads exert a pulling force on the actin filaments, causing the platelet plug to contract and shrink.
- Clot Stabilization: As the platelet plug contracts, the fibrin mesh becomes more tightly packed and stable, reinforcing the clot structure.
Significance of Clot Retraction
- Wound Closure: Clot retraction helps pull the wound edges closer together, promoting healing and reducing the risk of infection.
- Hemostasis: By reducing the size of the clot, clot retraction helps to limit blood loss.
- Vascular Patency: In small vessels, clot retraction can facilitate the reopening of the vessel lumen, restoring blood flow.
- Interplay with Fibrin Formation from Coagulation Cascade: Clot retraction compresses the fibrin mesh, making it denser and more resistant to breakdown. This stabilization helps to prolong the life of the clot and prevent rebleeding.
- Feedback Loop: The process of clot retraction can also stimulate the release of tissue factor from damaged tissue, further activating the coagulation cascade and reinforcing the clot.
Disorders of Primary Hemostasis
Quantitative Abnormalities of Platelets
Thrombocytopenia, defined as a platelet count below 150,000 per microliter (µL), can arise from three main mechanisms: decreased production, increased destruction, or sequestration. Each mechanism has a variety of underlying disorders, impacting the delicate balance of platelet production and survival impacting primary hemostasis.
1. Decreased Production
- Bone marrow disorders
- Aplastic anemia: Bone marrow failure leads to insufficient production of all blood cells, including platelets.
- Myelodysplastic syndromes (MDS): Abnormal blood cell development in the bone marrow can lead to reduced platelet production.
- Leukemia: Can infiltrate and damage the bone marrow, suppressing platelet production.
- Infiltration of bone marrow
- Cancer: Can infiltrate the bone marrow and replace healthy cells, including those responsible for platelet production.
- Fibrosis: Scarring of the bone marrow can reduce the space available for healthy cell production, including platelets.
- Nutritional deficiencies
- Vitamin B12 and folate deficiencies can impair platelet production as they are crucial for DNA synthesis and cell division.
- Medications: Certain drugs, such as chemotherapy agents, can suppress bone marrow function and reduce platelet production.
2. Increased Destruction
- Immune thrombocytopenia (ITP): The immune system mistakenly attacks and destroys platelets. This can be idiopathic (cause unknown) or secondary to infections, drugs, or autoimmune diseases.
- Heparin-induced thrombocytopenia (HIT): A rare reaction to heparin medication, where antibodies form against platelets, leading to their destruction.
- Evans syndrome: Autoimmune disorder targeting platelets and red blood cells, leading to their destruction.
- Disseminated intravascular coagulation (DIC): Widespread clotting throughout the body consumes platelets, leading to a low count.
- Sepsis: Severe bacterial infection can trigger widespread inflammation and platelet activation, leading to their destruction.
- Hemolytic uremic syndrome (HUS): Abnormal activation of the complement system destroys red blood cells and platelets.
- Thrombotic thrombocytopenic purpura (TTP): Deficiency in ADAMTS-13 which is essential for breaking down large protein molecules leads to platelet clumping and destruction.
- Drugs: Certain medications, such as antibiotics, can induce platelet destruction.
3. Sequestration
- Splenomegaly (enlarged spleen): The spleen can trap and destroy platelets, leading to a low count. This can be caused by various conditions like liver cirrhosis, portal hypertension, or certain infections.
- Liver cirrhosis: Scarring of the liver can increase pressure in the portal vein, leading to sequestration of platelets in the spleen.
- Hypersplenism: Functional overactivity of the spleen, causing it to destroy red blood cells and platelets.
- Medications: Some drugs can increase platelet sequestration in the spleen.
Qualitative Abnormalities of Platelets
While platelet count plays a crucial role in primary hemostasis, the quality of those platelets also matters. Inherited or acquired defects in platelet adhesion, activation, or platelet aggregation can lead to qualitative platelet dysfunction, hindering their ability to form effective clots in primary hemostasis and stop the bleeding.
1. Platelet Adhesion Defects
- Bernard-Soulier syndrome: A rare genetic disorder characterized by a deficiency in the GPIb/IX complex, a key receptor for platelets to adhere to collagen. This results in delayed and weak platelet binding, leading to prolonged bleeding.
- Glanzmann thrombasthenia: Another rare genetic disorder causing defective platelet adhesion due to mutations in the GPIIb/IIIa receptor, essential for fibrinogen binding and platelet aggregation. Symptoms mimic Bernard-Soulier syndrome with prolonged bleeding.
- Von Willebrand disease (VWD): A spectrum of disorders affecting vWF, a protein crucial for platelet adhesion to collagen and aggregation. Different types of VWD manifest with varying degrees of bleeding tendency.
2. Platelet Activation Defects
- Storage pool disorders: A group of rare genetic disorders characterized by deficiencies in platelet granules, leading to impaired release of essential contents like ADP, serotonin, and calcium ions. This results in weak platelet activation and aggregation, causing bleeding.
- P2Y12 receptor defects: Mutations in the P2Y12 receptor, critical for ADP signaling and platelet activation, can lead to impaired clotting and increased bleeding risk. This can be inherited or acquired due to medications like clopidogrel.
- Cyclooxygenase (COX) deficiency: Aspirin irreversibly inhibits COX, responsible for thromboxane A2 production, a potent platelet activator. This can cause temporary platelet dysfunction and increased bleeding risk.
3. Platelet Aggregation Defects
- Chediak-Higashi syndrome: A rare genetic disorder where abnormal granules within platelets lead to defective granule fusion and impaired platelet aggregation. This results in prolonged bleeding tendency.
- Wiskott-Aldrich syndrome (WAS): A rare genetic disorder characterized by a defective actin cytoskeleton in platelets, hindering their shape change and platelet aggregation ability. This leads to increased bleeding risk.
- Macrothrombocytopenia: A condition with large platelets due to abnormal granule distribution. While platelet count might be normal, their platelet aggregation function can be impaired, leading to bleeding.
Vascular Wall Abnormalities
The smooth lining of our blood vessels, the endothelium, is not just a passive barrier. It plays an active role in maintaining healthy blood flow and preventing clotting.
Defects in the endothelium, the innermost lining of blood vessels, or the underlying subendothelial structures can disrupt vWF binding and platelet adhesion for primary hemostasis, leading to bleeding complications.
1. Endothelial Dysfunction
- Atherosclerosis: This chronic inflammatory disease thickens and stiffens arterial walls, reducing blood flow and impairing the endothelium’s ability to produce nitric oxide, a vasodilator. This weakens vWF binding and platelet adhesion for primary hemostasis, potentially leading to bleeding.
- Thrombotic thrombocytopenic purpura (TTP): A rare disorder where a deficiency in ADAMTS13 enzyme leads to accumulation of large, multimeric vWF molecules. These can bind platelets indiscriminately, causing widespread clotting and microthrombi, paradoxically leading to bleeding in areas with poor circulation.
- Hemolytic uremic syndrome (HUS): This condition involves abnormal activation of the complement system, damaging endothelial cells and releasing pro-inflammatory factors. This can disrupt vWF binding and platelet adhesion, leading to bleeding and microthrombi formation.
- Sepsis: Severe bacterial infection triggers widespread inflammation, damaging the endothelium and reducing vWF expression. This can impair platelet adhesion and contribute to bleeding complications.
- Von Willebrand disease type IIB: This rare subtype of VWD arises from defective synthesis or release of vWF by endothelial cells, leading to insufficient vWF availability for platelet adhesion.
2. Subendothelial Defects
- Ehlers-Danlos syndrome (EDS): This group of genetic disorders affects collagen structure and strength within the vessel wall. This can lead to weakened subendothelial support, hindering vWF binding and platelet adhesion, and potentially causing bleeding.
- Antiphospholipid syndrome (APS): This autoimmune disorder involves antibodies targeting negatively charged phospholipids on cell surfaces, including those in the endothelium. This can disrupt vWF binding and platelet adhesion, leading to both thrombosis and bleeding complications.
- Henoch-Schönlein purpura (HSP): This vasculitic disorder damages small blood vessels, exposing the subendothelial matrix and triggering unwanted platelet adhesion and microthrombosis.
3. Acquired Endothelial Injuries
- Burns: Extensive burns can damage the endothelium, leading to impaired vWF binding and platelet adhesion, contributing to bleeding and delayed wound healing.
- Radiation therapy: Radiation can damage the endothelium and underlying structures, impacting vWF production and platelet adhesion. This can increase bleeding risk and impair wound healing.
- Certain medications: Some drugs, like aspirin or antiplatelet medications, can inhibit vWF function or platelet activation, temporarily affecting their binding and adhesion.
4. Inherited Connective Tissue Disorders
- Marfan syndrome: This genetic disorder affects connective tissue throughout the body, including blood vessels, potentially leading to vascular fragility and impaired platelet adhesion.
Common Symptoms and Clinical Presentation of Primary Hemostasis Disorders
Primary hemostasis disorders primarily affect the initial formation of the platelet plug, leading to characteristic bleeding patterns. The common symptoms and clinical presentations often reflect this impaired ability to stop bleeding from superficial injuries or small blood vessels.
Mucocutaneous Bleeding
This is the hallmark of primary hemostasis disorders. Bleeding occurs from mucous membranes and the skin.
- Petechiae: These are tiny, pinpoint, non-blanching red or purple spots on the skin, often appearing on the lower legs. They result from small hemorrhages from capillaries. Petechiae are highly suggestive of a platelet disorder.
- Purpura/Ecchymoses: These are larger bruises (purpura are generally smaller than 1 cm, while ecchymoses are larger). They are often spontaneous or occur with minimal trauma, and can be widespread.
- Epistaxis (Nosebleeds): Frequent, prolonged, or difficult-to-stop nosebleeds are a very common symptom.
- Gingival Bleeding (Gum Bleeding): Bleeding from the gums, especially during brushing or spontaneously, is another frequent complaint.
- Menorrhagia (Heavy Menstrual Bleeding): In women, unusually heavy, prolonged, or frequent menstrual periods are a significant indicator.
- Gastrointestinal Bleeding: While less common than mucocutaneous bleeding, GI bleeding can occur, manifesting as blood in stool (melena or hematochezia) or vomit (hematemesis).
- Hematuria (Blood in Urine): Blood in the urine can also be a symptom, though less common than other mucocutaneous bleeding.
Immediate Bleeding After Injury or Surgery
Unlike secondary hemostasis disorders (which involve clotting factor deficiencies and often present with delayed bleeding), primary hemostasis disorders typically cause bleeding that starts immediately after a cut, scrape, or surgical procedure (like dental extractions) and can be prolonged.
Superficial Bleeding
The bleeding tends to be superficial, meaning it’s often visible on the skin or from mucous membranes, rather than deep internal bleeding into joints (hemarthrosis) or muscles (hematomas), which are more characteristic of secondary hemostasis disorders.
General Laboratory Investigations of Primary Hemostasis Disorders
- Complete Blood Count (CBC) with Platelet Count: The platelet count is particularly important in evaluating primary hemostasis, as a low count (thrombocytopenia) can directly impact platelet plug formation. The CBC can also reveal other abnormalities like anemia or elevated white blood cell counts (leukocytosis) that might hint at underlying conditions affecting primary hemostasis.
- Peripheral Blood Smear Examination: This examination allows direct visualization of platelet morphology and size under a microscope. Abnormalities in platelet size or shape (macroplatelets, microplatelets, giant platelets) can indicate specific disorders like Bernard-Soulier syndrome or Wiskott-Aldrich syndrome. The smear also helps confirm the platelet count obtained in the CBC.
- Bleeding Time Test: This test assesses platelet adhesion and platelet aggregation at the site of injury. It measures the time it takes for a small standardized wound to stop bleeding. A prolonged bleeding time suggests impaired platelet function disrupting primary hemostasis, although it’s not a specific test and can be influenced by other factors like skin thickness or blood flow.
- Specific Assays for vWF Levels and Function: Von Willebrand factor (vWF) plays a critical role in platelet adhesion to the injured vessel wall. Measuring vWF antigen levels can identify quantitative deficiencies, but additional tests are needed to assess its functionality. Assays for ristocetin cofactor activity (vWF:RCo) and collagen-binding affinity help determine if vWF is working properly, further pinpointing potential causes of bleeding.
- Platelet Aggregation Studies: These tests analyze the ability of platelet aggregation in response to various stimuli like ADP, collagen, or adrenaline. They can distinguish between platelet adhesion defects from those affecting platelet activation and platelet aggregation. Different platelet aggregation patterns can point towards specific disorders like Glanzmann thrombasthenia or storage pool disorders.
- Genetic Testing: For disorders with known genetic mutations, like Bernard-Soulier syndrome, Wiskott-Aldrich syndrome, or some types of VWD, genetic testing can provide definitive diagnosis and help with understanding the inheritance pattern and future risks.
Managing Primary Hemostasis Disorders
Treating primary hemostasis disorders requires a multifaceted approach, aimed at addressing the underlying cause while preventing or minimizing bleeding symptoms.
- Addressing the Underlying Cause
- Infections: Treating underlying infections associated with thrombocytopenia, like viral hepatitis or Epstein-Barr virus, can help resolve platelet count issues.
- Nutritional deficiencies: Correcting deficiencies in vitamin B12, folate, or iron, which are essential for platelet production, can improve platelet count.
- Medications: Discontinuing medications that may be causing thrombocytopenia due to drug-induced immune reactions is crucial.
- Autoimmune disorders: Immunosuppressive therapy can be used to manage autoimmune conditions like ITP, reducing platelet destruction.
- Cancer: Treating underlying malignancies can improve platelet production in cases of bone marrow infiltration.
- Platelet Transfusion: For severe thrombocytopenia with active bleeding or high bleeding risk, platelet transfusions can provide the necessary platelets for effective clot formation in primary hemostasis. This is an important temporary measure to control bleeding until the underlying cause is addressed or platelet production recovers.
- Antifibrinolytic Drugs: Medications like aminocaproic acid or tranexamic acid prevent the breakdown of fibrin within the existing platelet plug by inhibiting plasmin, the enzyme responsible for fibrinolysis. This helps stabilize the clot in primary hemostasis and reduce bleeding in various situations, including menorrhagia or epistaxis.
- Desmopressin: This synthetic hormone can stimulate the release of vWF from endothelial cells, potentially improving platelet adhesion and reducing bleeding in mild cases of von Willebrand disease. It’s a non-invasive and readily available option for managing bleeding during procedures or episodes.
- Bone Marrow Transplantation: In severe inherited platelet disorders like Wiskott-Aldrich syndrome, bone marrow transplantation can offer a potential cure by replacing the defective bone marrow with healthy donor cells. This is a complex and potentially risky procedure, requiring careful consideration of the benefits and risks.
Frequently Asked Questions (FAQs)
What is the difference between primary and secondary bleeding disorders?
The main difference between primary and secondary bleeding disorders lies in which part of the blood clotting cascade is affected.
- Primary bleeding disorders: These involve problems with platelet function. Platelets are the first responders in clotting, forming a plug at the site of injury. If platelets are dysfunctional or deficient, the initial plug formation is impaired, leading to easy bruising and bleeding from minor injuries. Examples include Glanzmann’s disease and Bernard-Soulier syndrome.
- Secondary bleeding disorders: These involve problems with the coagulation factors. These are proteins in the blood plasma that work together in a cascade to form a stronger fibrin clot to solidify the initial platelet plug. Deficiencies or abnormalities in these factors prevent the formation of a stable clot, leading to deeper and more prolonged bleeding episodes. Examples include hemophilia A and B, and von Willebrand disease.
| Feature | Primary Bleeding Disorders | Secondary Bleeding Disorders |
|---|---|---|
| Affected part of clotting | Platelet function | Coagulation factors (plasma proteins) |
| Symptoms | Easy bruising, bleeding from minor injuries, mucous membrane bleeding | Deep muscle bleeds, prolonged bleeding after surgery or injury, internal bleeding |
| Examples | Glanzmann’s disease, Bernard-Soulier syndrome | Hemophilia A, B, von Willebrand disease |
What is the difference between primary and secondary platelet aggregation?
Primary platelet aggregation refers to the initial, reversible clumping of platelets at the site of vascular injury in primary hemostasis. This process is primarily driven by the binding of platelets to von Willebrand factor (vWF) and exposed collagen in the damaged vessel wall.
This initial adhesion and weak platelet aggregation form the first layer of the platelet plug. The platelet aggregation at this stage is largely due to mechanical forces and the initial release of small amounts of ADP and thromboxane A2 (TxA2) from activated platelets.
Secondary aggregation follows primary platelet aggregation and involves a more robust and irreversible clumping of platelets, leading to the formation of a stable, definitive platelet plug.
What is the role of fibrinogen in primary hemostasis?
In primary hemostasis, fibrinogen, a soluble plasma protein, plays a critical role by acting as a crucial bridging molecule between activated platelets. Once platelets are activated at the site of injury, they undergo a conformational change, exposing their glycoprotein IIb/IIIa (GPIIb/IIIa) receptors.
Fibrinogen then binds to these activated receptors on adjacent platelets, forming strong cross-links that facilitate the robust and irreversible platelet aggregation, which is essential for the formation of a stable and definitive platelet plug in primary hemostasis.
Disclaimer: This article is intended for informational purposes only and is specifically targeted towards medical students. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Saba HI, Roberts HR. Hemostasis and Thrombosis: Practical Guidelines in Clinical Management (Wiley Blackwell). 2014.
- DeLoughery TG. Hemostasis and Thrombosis 4th Edition (Springer). 2019.
- Keohane EM, Otto CN, Walenga JM. Rodak’s Hematology 6th Edition (Saunders). 2019.
- Kaushansky K, Levi M. Williams Hematology Hemostasis and Thrombosis (McGraw-Hill). 2017.
- McRae S. Physiological Haemostasis. In: Fitridge R, Thompson M, editors. Mechanisms of Vascular Disease: A Reference Book for Vascular Specialists [Internet]. Adelaide (AU): University of Adelaide Press; 2011. 9. Available from: https://www.ncbi.nlm.nih.gov/books/NBK534253/
- LaPelusa A, Dave HD. Physiology, Hemostasis. [Updated 2023 May 1]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK545263/
- Gale AJ. Continuing Education Course #2: Current Understanding of Hemostasis. Toxicologic Pathology. 2010;39(1):273-280. doi:10.1177/0192623310389474
- Godby RC, May JE, Lima JLO, Singh N, Marques MB. Congenital and acquired disorders of primary hemostasis. Ann Blood 2022;7:10.
- Scridon, A. (2022). Platelets and Their Role in Hemostasis and Thrombosis—From Physiology to Pathophysiology and Therapeutic Implications. International Journal of Molecular Sciences, 23(21), 12772. https://doi.org/10.3390/ijms232112772