Key Takeaways
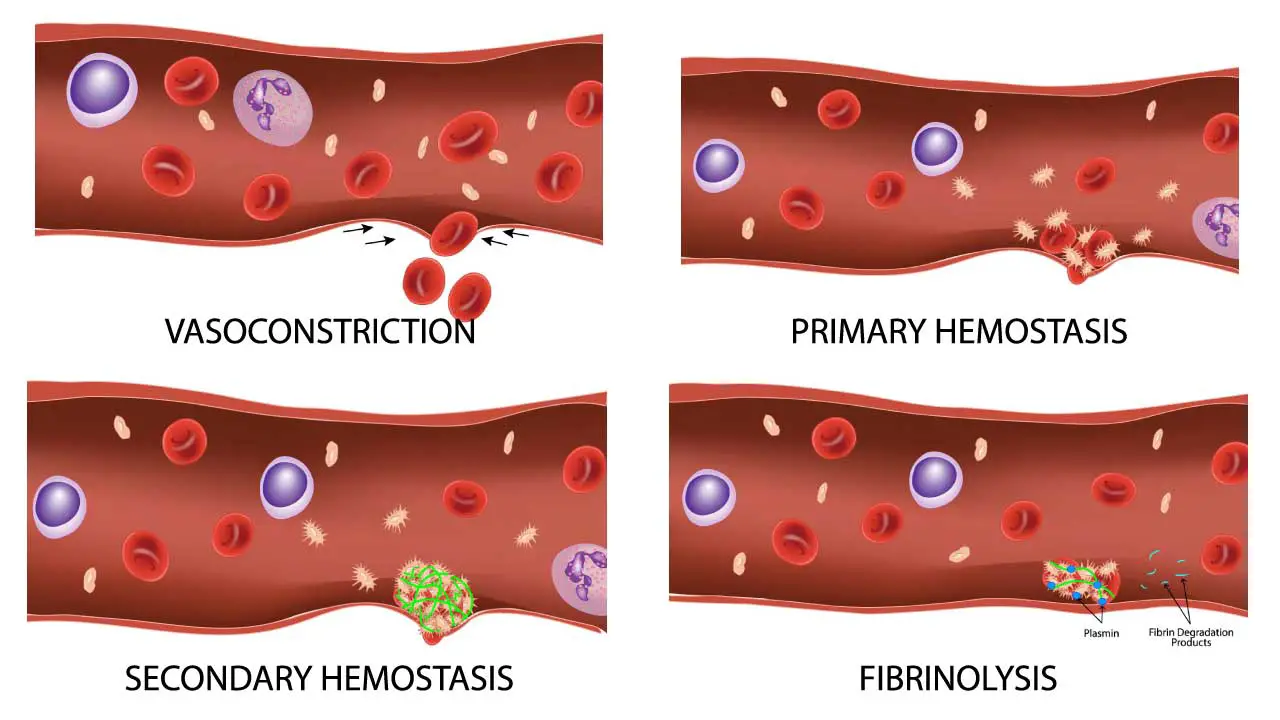
Hemostasis is the body's coordinated system for stopping bleeding while keeping blood flowing inside healthy vessels. It works in four overlapping stages: vasoconstriction, primary hemostasis, secondary hemostasis, and fibrinolysis.
- Vasoconstriction ▾: The immediate constriction of injured blood vessels by the surrounding smooth muscle helps reduce blood flow to the wound, minimizing initial blood loss. This buys precious time for the next stages to kick in, and its effectiveness influences the amount of blood platelets needed to adhere to.
- Plug formation ▾: Primary hemostasis forms the initial platelet plug. Platelets stick to exposed collagen through von Willebrand factor and the GPIb receptor, then aggregate via GPIIb/IIIa receptors that bind fibrinogen.
- Coagulation Cascade ▾: Secondary hemostasis produces fibrin, the protein mesh that stabilizes the plug. The cell-based model describes this as three phases on cell surfaces: initiation, amplification, and propagation. The intrinsic/extrinsic cascade still explains laboratory tests like PT and aPTT.
- Fibrinolysis ▾: Once the wound starts to heal, plasmin, an enzyme, breaks down fibrin, dissolving the clot and allowing blood flow to resume.
- Potential Problems ▾: Failure of hemostasis leads to two opposite problems: bleeding disorders (hemophilia, von Willebrand disease, thrombocytopenia) and thrombotic disorders (DVT, pulmonary embolism, stroke).
*Click ▾ for more information
Why Hemostasis Matters
Picture a child with hemophilia whose knee swells painfully every few weeks from a tiny internal bleed, or a postoperative patient who develops a clot in a leg vein after surgery. Both situations come down to the same biological balance: hemostasis. When this system works, you barely notice it. When it fails in either direction, the consequences range from bruising and joint damage to stroke and death.
Hemostasis is the process that keeps blood inside vessels and stops bleeding when those vessels are injured [1]. It has to be fast, local, and reversible — clot where you must, but not where you shouldn't.
The mechanism unfolds in four overlapping stages: vasoconstriction, primary hemostasis, secondary hemostasis, and fibrinolysis. The endothelium (the inner lining of blood vessels) sets the stage by deciding when clotting starts and stops.
The Endothelium
Before talking about clots, it helps to know what stops them from forming all the time. Healthy endothelial cells line every blood vessel and constantly release substances that keep platelets calm and clotting factors in check [4]:
- Nitric oxide (NO) and prostacyclin (PGI₂) stop platelets from sticking and relax smooth muscle.
- Thrombomodulin binds thrombin and converts it from a clot-promoter into a clot-suppressor by activating protein C.
- Tissue factor pathway inhibitor (TFPI) dampens early clotting signals.
- Heparan sulfate on the endothelial surface activates antithrombin, which neutralizes thrombin and factor Xa.
Hemostasis begins only when this protective layer is damaged. Once collagen and tissue factor are exposed, the four-stage process kicks in.
Stage 1: Vasoconstriction
The first response to vessel injury is reflex narrowing. Smooth muscle in the vessel wall contracts, reducing blood flow to the wound and limiting initial blood loss. Several signals drive this:
- Mechanical stretch on the smooth muscle from the injury itself.
- Thromboxane A₂ released by activated platelets.
- Endothelin-1 released by injured endothelial cells.
Vasoconstriction is brief but buys time for platelets and clotting factors to arrive.
Stage 2: Primary Hemostasis
Primary hemostasis builds a soft, temporary seal at the injury site. It happens in three connected steps: adhesion, activation, and aggregation [1].
Platelets at a Glance
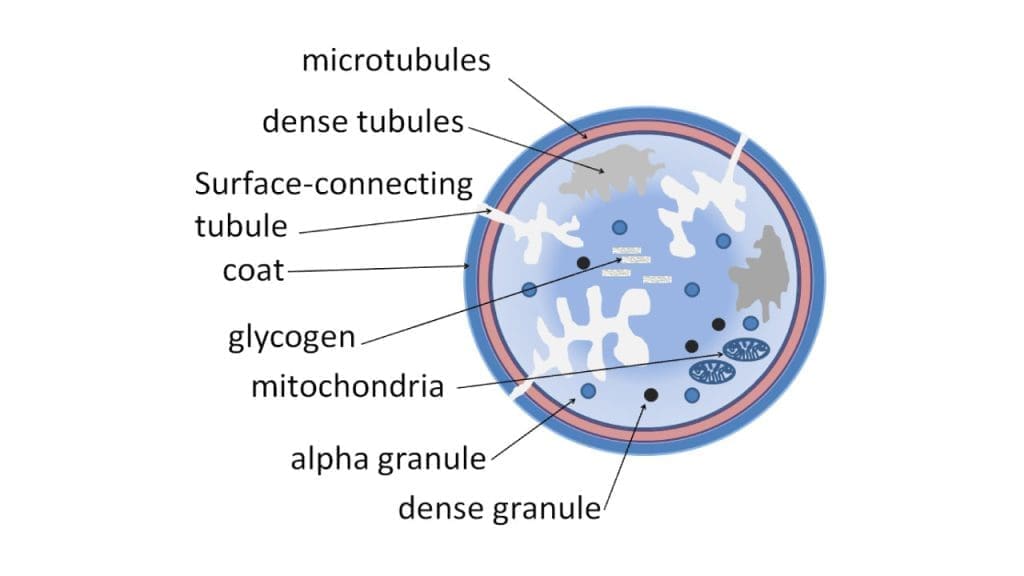
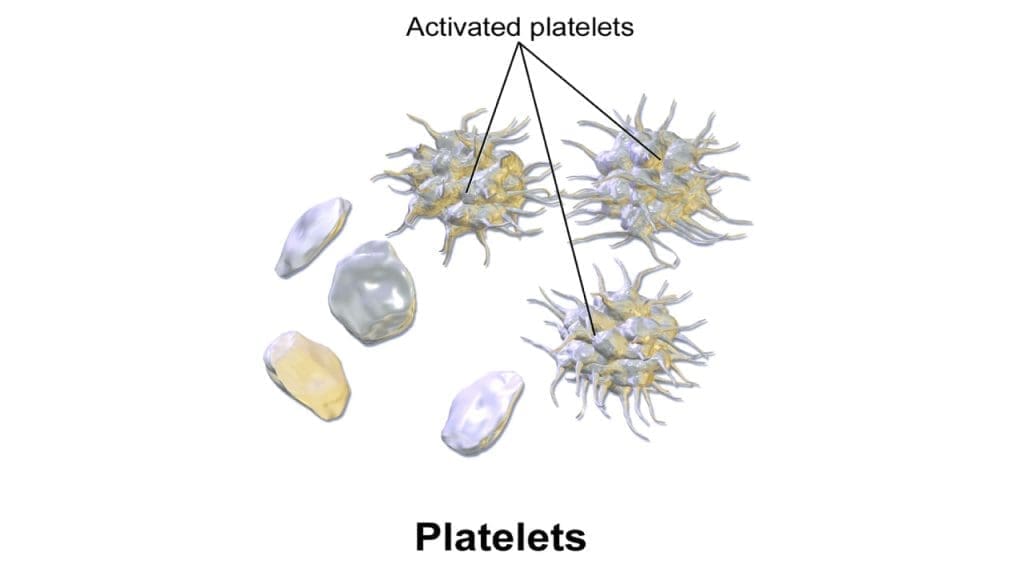
Platelets are small, disc-shaped cell fragments derived from megakaryocytes in the bone marrow. They have no nucleus but are packed with granules and signaling machinery. A normal count is 150–450 × 10⁹/L. Their surface carries the receptors that drive every step below.
Adhesion: Anchoring to the Wound
When endothelium is breached, collagen and von Willebrand factor (vWF) in the subendothelial matrix become exposed. Under flowing blood, especially in arteries where shear is high, vWF acts as a bridge: it binds collagen on one side and the platelet receptor glycoprotein Ib (GPIb) on the other [4]. This vWF–GPIb tether catches platelets out of the bloodstream like a rope catching a passing swimmer. Once tethered, platelets bind collagen directly through other receptors (GPVI, integrin α2β1) and stop rolling.
This step explains why von Willebrand disease produces a primary hemostasis defect even when platelet count is normal.
Activation: Switching Platelets On
Anchored platelets change shape from smooth discs into spiky spheres, which dramatically increases their surface area. They release the contents of their granules:
- Dense granules release ADP, ATP, serotonin, and calcium.
- Alpha granules release vWF, fibrinogen, factor V, and growth factors.
They also synthesize and release thromboxane A₂ through cyclooxygenase-1 (COX-1). ADP and thromboxane A₂ are powerful recruitment signals — they bring more platelets to the site and activate them too.
Effects of aspirin
Aspirin irreversibly inhibits platelet COX-1. Because platelets cannot make new COX-1 (no nucleus), the effect lasts the lifespan of the platelet (roughly 7–10 days). This is why aspirin is held for about a week before elective surgery.
Aggregation: Building the Plug
Activation reshapes the platelet receptor GPIIb/IIIa (integrin αIIbβ3) from a low-affinity state to a high-affinity one. Activated GPIIb/IIIa binds fibrinogen, which has two binding ends. One fibrinogen molecule can therefore bridge two platelets, and as more platelets pile in, a dense plug forms.
This plug is enough to stop minor bleeding on its own, but it is mechanically fragile. To make it last, the body needs fibrin.
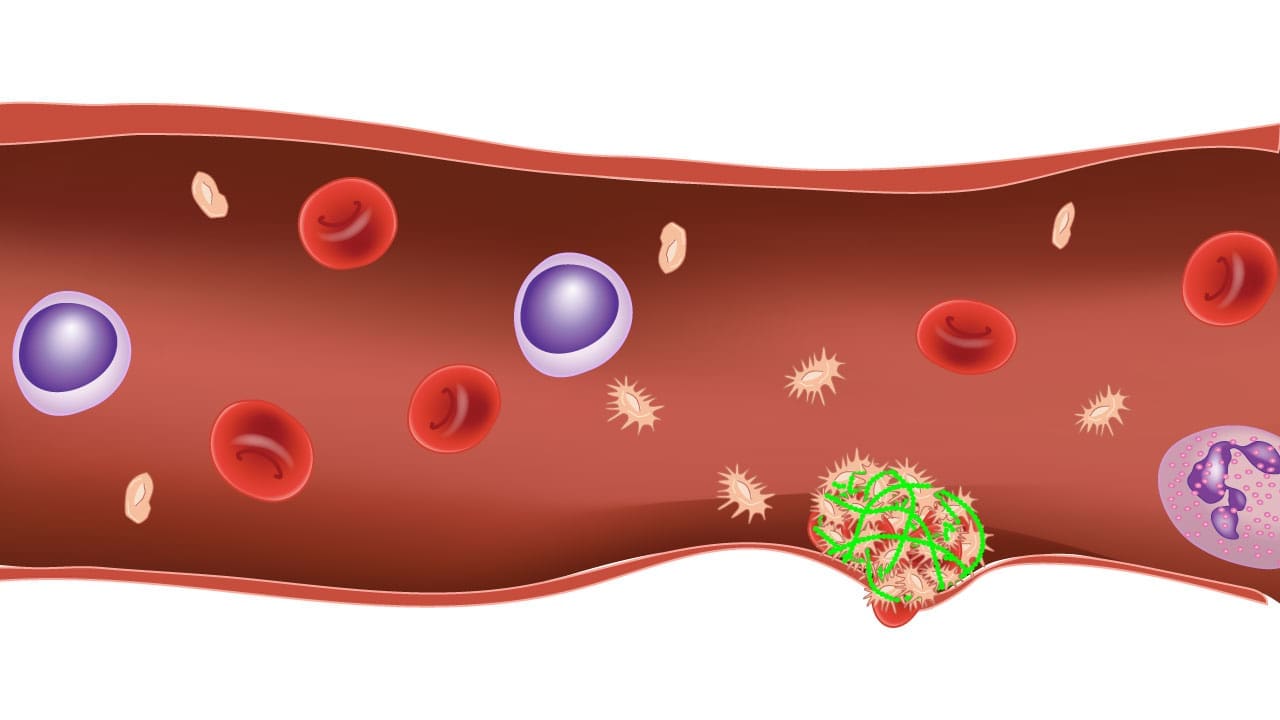
Stage 3: Secondary Hemostasis
Secondary hemostasis converts the soluble plasma protein fibrinogen into an insoluble fibrin mesh that locks the platelet plug in place. The pathway is named the coagulation cascade because each step activates the next.
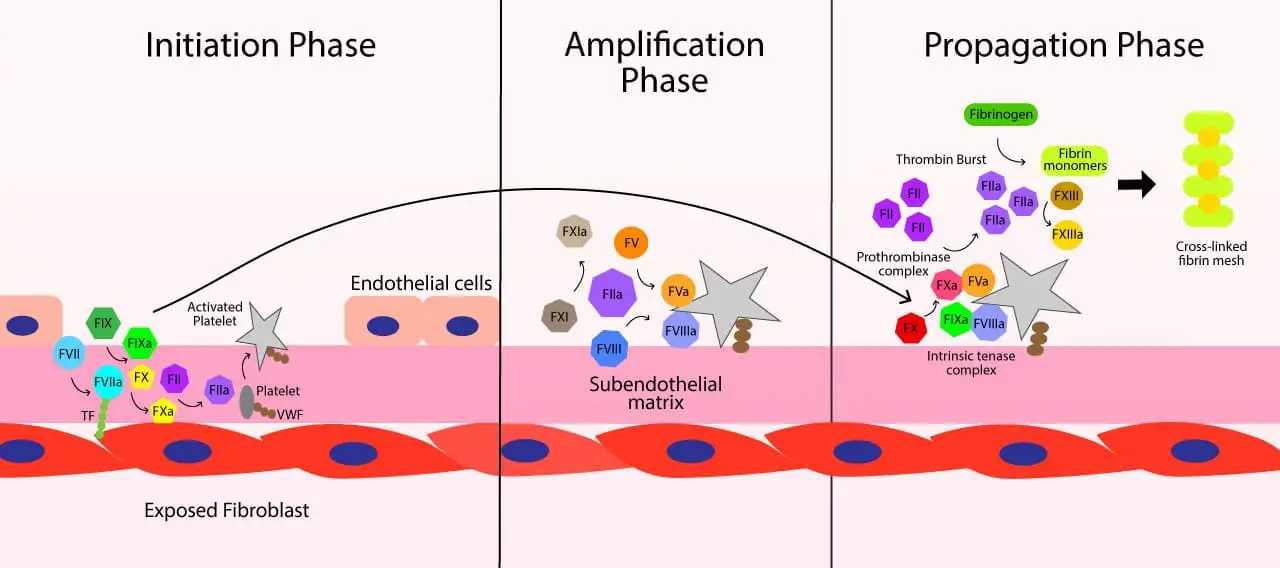
The Cell-Based Model: How It Actually Happens In Vivo
The traditional intrinsic/extrinsic/common pathway diagram is useful for interpreting lab tests, but it does not match what happens inside a living blood vessel. The accepted physiological model since 2001 is the cell-based model, with three overlapping phases on cell surfaces [2,3]:
- Initiation — Tissue factor (TF), exposed on subendothelial cells when the vessel is injured, binds factor VIIa. The TF/VIIa complex activates a small amount of factors IX and X, generating a tiny burst of thrombin. This burst is too small to clot blood on its own.
- Amplification — That trace thrombin is the trigger that activates platelets, factor V, factor VIII (by separating it from vWF), and factor XI. The activated platelet surface becomes the assembly platform for the next phase.
- Propagation — On the activated platelet surface, factor IXa (with VIIIa) generates more factor Xa, and factor Xa (with Va) drives a large thrombin burst. This burst converts fibrinogen to fibrin and activates factor XIII, which cross-links fibrin into a stable, insoluble mesh.
This model explains why hemophilia (deficiency of factor VIII or IX) causes bleeding even though the initiation step is intact: without VIII or IX, the propagation burst never happens, and clots stay fragile.
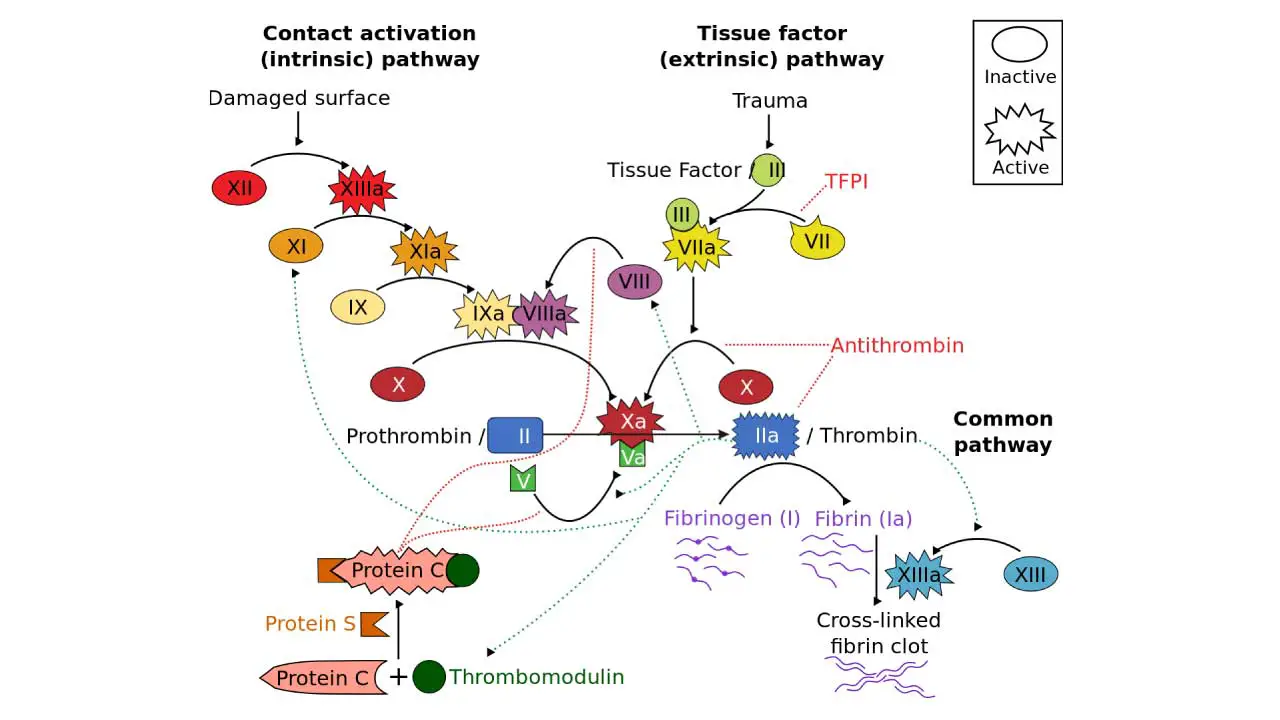
The Cascade Model: A Lab-Test Framework
The older cascade model splits coagulation into intrinsic and extrinsic pathways that converge on a common pathway. It does not reflect in vivo physiology, but it does map directly onto the lab tests every clinician orders:
- Extrinsic pathway (factors VII, X, V, II, fibrinogen) — measured by prothrombin time (PT) and reported as INR.
- Intrinsic pathway (factors XII, XI, IX, VIII, X, V, II, fibrinogen) — measured by activated partial thromboplastin time (aPTT).
- Common pathway — factors X, V, prothrombin (II), and fibrinogen (I).
A prolonged PT points to the extrinsic side: warfarin therapy, vitamin K deficiency, or liver disease. A prolonged aPTT points to the intrinsic side: heparin, hemophilia A or B, or a lupus anticoagulant. Both prolonged together suggests the common pathway, severe liver disease, or DIC.
Vitamin K's Role
Vitamin K is required for the liver to produce functional factors II, VII, IX, and X, plus the regulatory proteins C and S. Warfarin works by blocking vitamin K recycling, which is why it lowers all four factors over a few days. Newborns get a vitamin K injection at birth because they are functionally deficient.
Natural Anticoagulants
A clot that grew unchecked could fill the entire vascular tree. Several built-in brakes confine clotting to the wound [4]:
- Antithrombin inactivates thrombin and factor Xa. Heparin works by accelerating this reaction up to 1,000-fold.
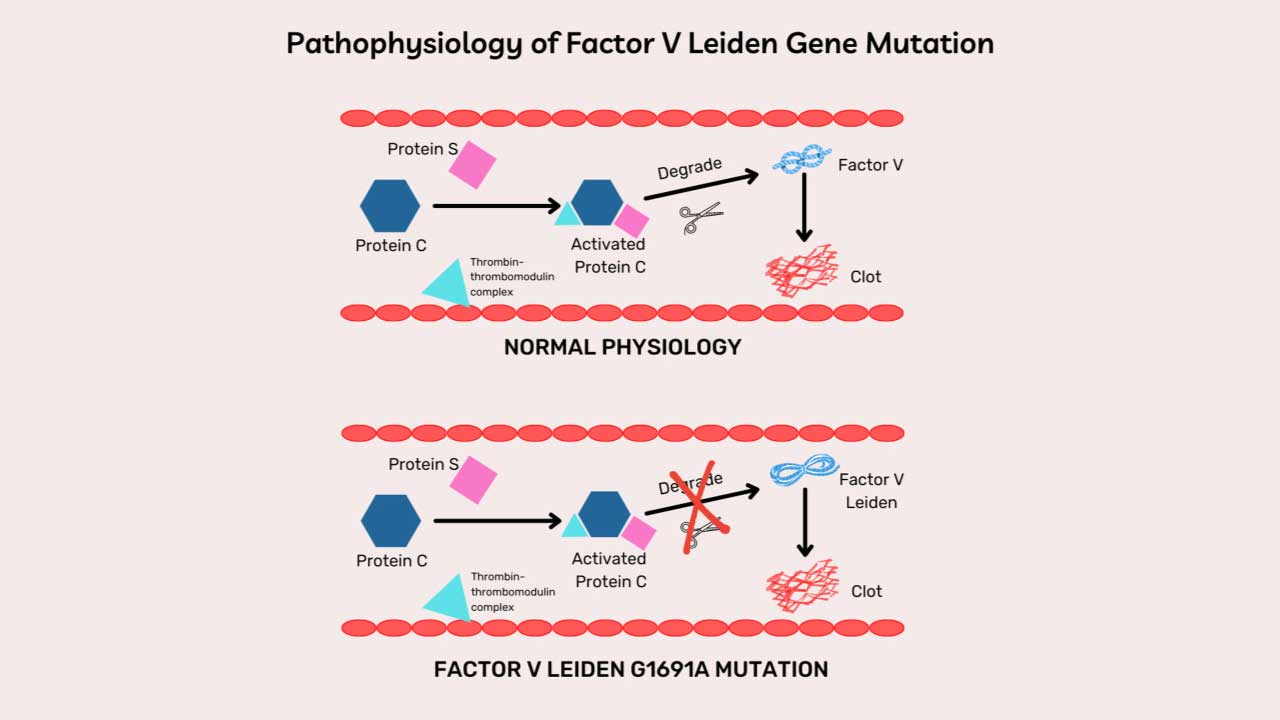
- Protein C and protein S — Thrombin bound to thrombomodulin (on intact endothelium) activates protein C, which, with protein S as a cofactor, inactivates factors Va and VIIIa.
- Tissue factor pathway inhibitor (TFPI) shuts down the TF/VIIa complex once propagation is underway.
Inherited deficiencies of any of these (antithrombin, protein C, protein S) cause thrombophilia — an increased clotting tendency. The most common inherited thrombophilia, Factor V Leiden, makes factor Va resistant to inactivation by activated protein C.
Stage 4: Fibrinolysis
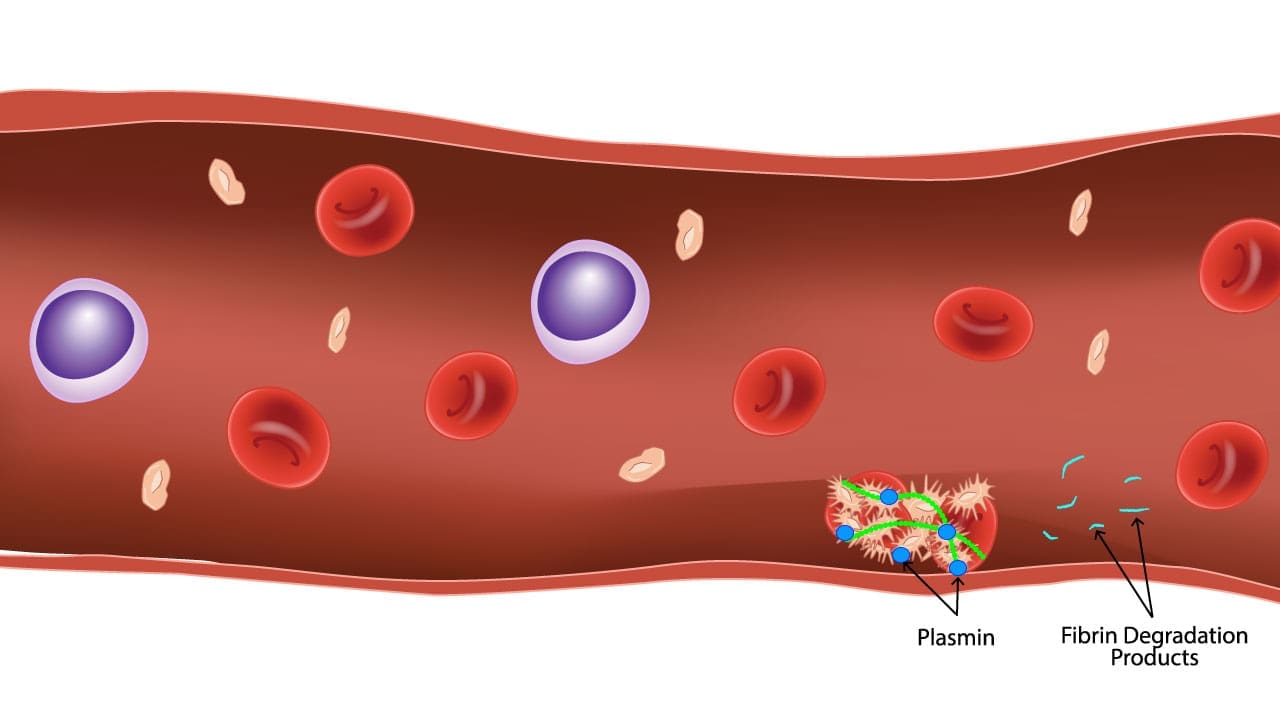
Once tissue has healed, the clot needs to come down. Fibrinolysis is the enzymatic breakdown of fibrin by plasmin.
The liver produces plasminogen, an inactive precursor that circulates in plasma. Tissue plasminogen activator (tPA), released by endothelial cells, converts plasminogen into active plasmin on the fibrin surface. Plasmin cuts fibrin into fragments, including D-dimer, which is detectable in blood and used as a clinical marker.
Plasmin is itself controlled. Plasminogen activator inhibitor-1 (PAI-1) blocks tPA, and α2-antiplasmin neutralizes free plasmin in the circulation. The system stays local: plasmin is most active where fibrin is, which is exactly where it's needed.
D-dimer: The rule out test
D-dimer is highly sensitive but not specific. It rises in DVT, PE, DIC, infection, recent surgery, and pregnancy. It is most useful as a rule-out test in patients with low pre-test probability for clots.
Hemostatic Disorders
Disruption of hemostasis at any stage produces clinical disease.
Bleeding Disorders
- Platelet disorders — Low count (thrombocytopenia from ITP, marrow failure, DIC) or impaired function (von Willebrand disease, Glanzmann thrombasthenia, Bernard-Soulier syndrome).
- Clotting factor deficiencies — Hemophilia A (factor VIII), hemophilia B (factor IX), and rarer factor deficiencies.
- Vascular disorders — Hereditary hemorrhagic telangiectasia, Ehlers-Danlos syndrome.
- Acquired causes — Liver disease, vitamin K deficiency, anticoagulant overdose.
Thrombotic Disorders
- Inherited thrombophilias — Factor V Leiden, prothrombin G20210A, deficiencies of antithrombin, protein C, protein S.
- Acquired thrombophilias — Antiphospholipid syndrome, malignancy, myeloproliferative neoplasms (often JAK2 mutation).
- Situational triggers — Surgery, immobility, pregnancy, hormonal therapy.
- Disseminated intravascular coagulation (DIC) — A consumptive process in which sepsis, malignancy, or obstetric emergencies trigger widespread clotting that uses up platelets and factors. The patient simultaneously clots and bleeds. Hallmarks: low platelets, prolonged PT/aPTT, low fibrinogen, very high D-dimer.
Recognizing Hemostatic Disorders Clinically
Pattern recognition helps narrow the cause before any lab test.
Bleeding Patterns
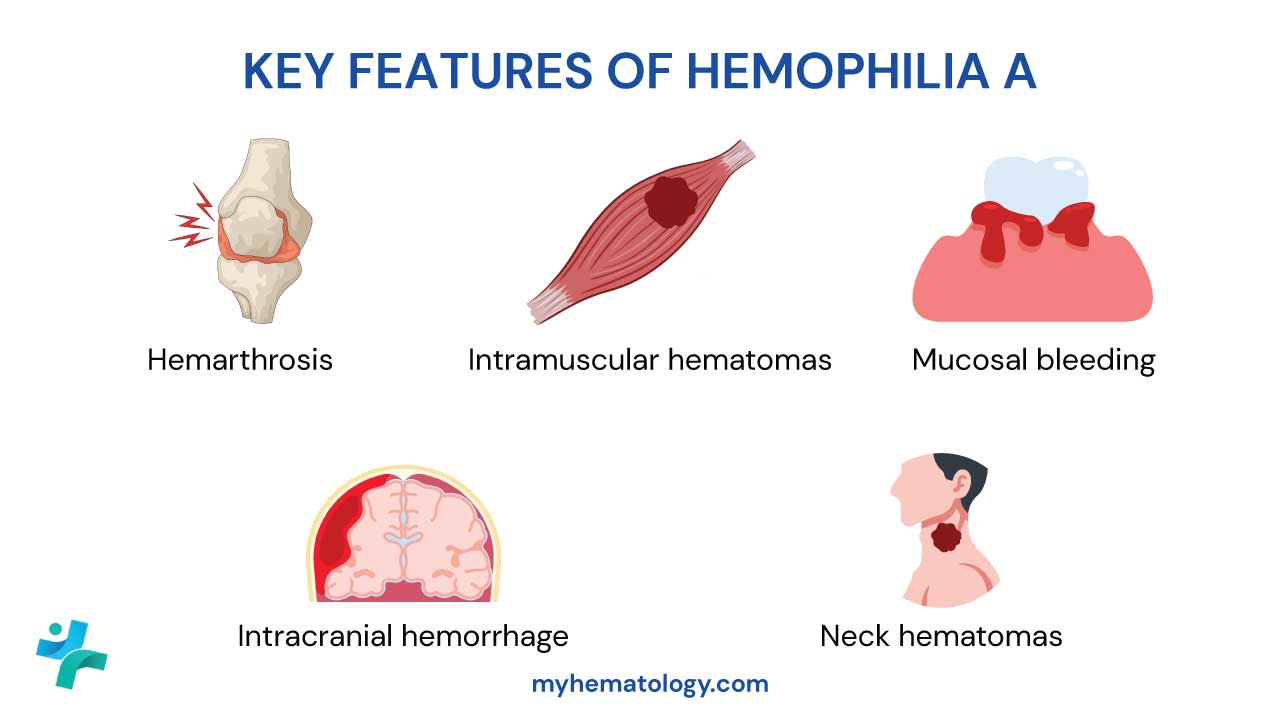
- Mucocutaneous bleeding — petechiae, purpura, nosebleeds, gum bleeding, heavy periods. Suggests a primary hemostasis problem (platelets or vWF).
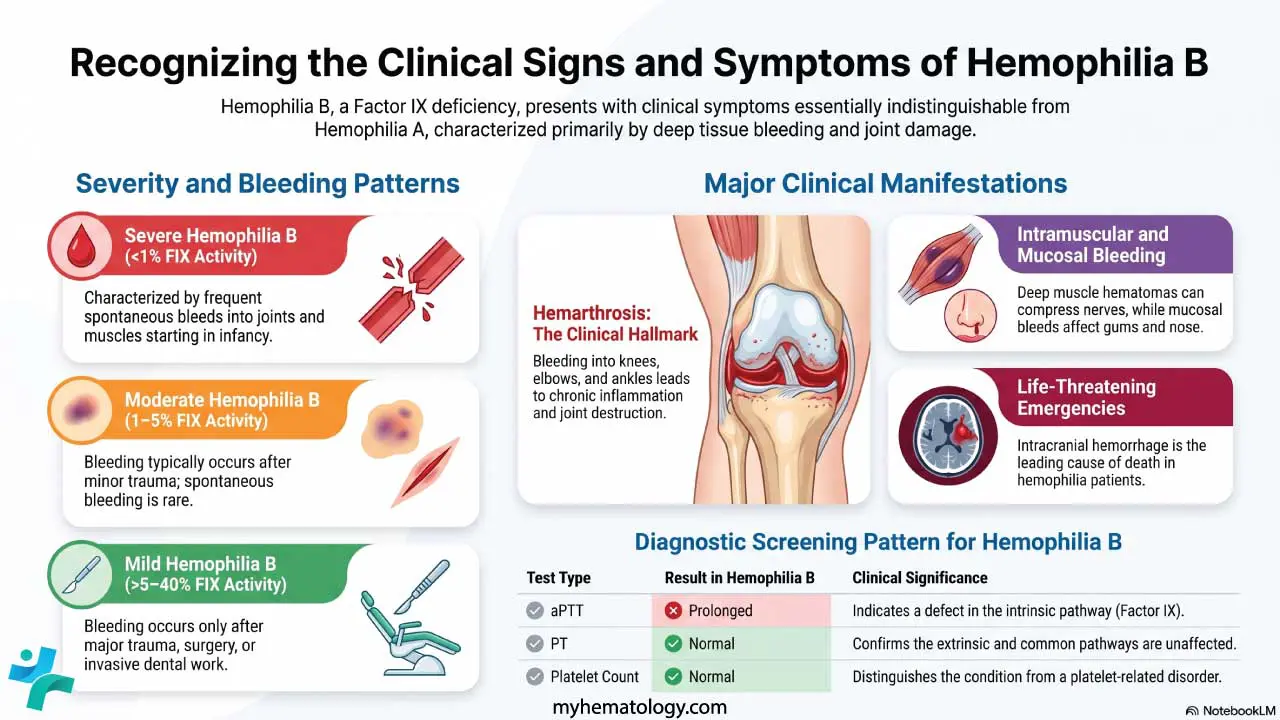
- Deep tissue bleeding — joint bleeds (hemarthrosis), large muscle hematomas, intracranial hemorrhage. Suggests a secondary hemostasis problem (clotting factors).
- Delayed bleeding after surgery or dental work — classic for factor deficiencies.
Thrombotic Patterns
- Venous thromboembolism (VTE) — Unilateral leg swelling and pain (DVT), sudden shortness of breath and pleuritic chest pain (PE).
- Arterial thrombosis — Myocardial infarction, ischemic stroke, limb ischemia.
Key History Questions
- Bleeding or clotting episodes — onset, frequency, severity, triggers.
- Family history of bleeding, clotting, or pregnancy loss.
- Medication review — anticoagulants, antiplatelets, NSAIDs.
- Comorbidities — liver disease, kidney disease, autoimmune disease, malignancy.
Laboratory Approach to Hemostasis
Initial screen:
- Complete blood count (CBC) with platelet count.
- PT/INR — extrinsic and common pathway.
- aPTT — intrinsic and common pathway.
- Fibrinogen and thrombin time (TT) — final step of coagulation.
Targeted follow-up depends on the screen:
- Specific factor assays (VIII, IX, XI, etc.) for isolated aPTT prolongation.
- vWF antigen, activity, and multimers for suspected von Willebrand disease.
- Platelet function assays (PFA-100, light transmission aggregometry) for qualitative platelet defects.
- D-dimer for VTE rule-out and DIC support.
- Thrombophilia panel (Factor V Leiden, prothrombin gene mutation, antithrombin, protein C, protein S, antiphospholipid antibodies) for unexplained or recurrent thrombosis.
A mixing study distinguishes factor deficiency (corrects with normal plasma) from an inhibitor like a lupus anticoagulant (does not correct).
Treatment Landscape
Treatment targets the specific defect.
For Bleeding Disorders
- Factor concentrates — recombinant factor VIII for hemophilia A, factor IX for hemophilia B. The mainstay of hemophilia care.
- Emicizumab — a bispecific antibody that mimics activated factor VIII by bridging factor IXa and X. Subcutaneous, weekly to monthly, and effective even in patients with inhibitors. Standard-of-care prophylaxis for hemophilia A [6].
- Non-factor "rebalancing" therapies — Rather than replacing missing clotting factors, these novel agents inhibit the body's natural anticoagulants to restore thrombin generation. Anti-TFPI monoclonal antibodies (e.g., marstacimab) and siRNA therapeutics (e.g., fitusiran, which silences antithrombin production in the liver) offer infrequent subcutaneous prophylaxis for hemophilia A and B, regardless of inhibitor status [11,12].
- Gene therapy — Valoctocogene roxaparvovec (hemophilia A) and etranacogene dezaparvovec (hemophilia B) deliver functional factor genes via AAV vectors. While they produced sustained factor levels in initial trials, real-world adoption has been significantly challenged by waning durability (factor levels dropping over time), high costs, and market withdrawals, shifting the view of gene therapy from a definitive cure to a complex, individualized option [7,8,13].
- Desmopressin (DDAVP) — releases stored vWF and factor VIII from endothelium, useful in mild hemophilia A and type 1 von Willebrand disease.
- Tranexamic acid — blocks plasminogen activation, reducing fibrinolysis. Used in heavy menstrual bleeding, dental procedures, trauma, and postpartum hemorrhage.
- Platelet transfusion — for severe thrombocytopenia or platelet function disorders with active bleeding.
- Caplacizumab — an anti-vWF nanobody that blocks platelet–vWF interaction. Used in acquired TTP alongside plasma exchange and immunosuppression [9].
For Thrombotic Disorders
- Heparin (unfractionated and low-molecular-weight) — accelerates antithrombin's inhibition of thrombin and factor Xa.
- Warfarin — inhibits vitamin K-dependent factors (II, VII, IX, X). Monitored with INR.
- Direct oral anticoagulants (DOACs) — dabigatran (anti-thrombin); apixaban, rivaroxaban, edoxaban (anti-factor Xa). Preferred over warfarin for most VTE and atrial fibrillation indications.
- Antiplatelet drugs — aspirin (COX-1 inhibitor), clopidogrel/ticagrelor/prasugrel (P2Y12 inhibitors). Used mainly for arterial disease.
- Thrombolytics — tPA, tenecteplase. Reserved for life- or limb-threatening clots (large PE, acute stroke, STEMI).
Reversing Anticoagulants in an Emergency
When a patient on anticoagulation bleeds critically or needs urgent surgery, reversal is sometimes essential [5]:
- Vitamin K + 4-factor PCC for warfarin.
- Idarucizumab for dabigatran (a monoclonal antibody fragment that binds dabigatran directly).
- 4-factor prothrombin complex concentrate (4F-PCC) or Andexanet alfa are utilized to reverse the Factor Xa inhibitors apixaban and rivaroxaban. While andexanet alfa is a specifically designed recombinant decoy, many institutions rely on 4F-PCC. Recent systematic reviews and meta-analyses demonstrate that andexanet alfa does not provide a statistically significant advantage over 4F-PCC in reducing short-term mortality and is associated with an increased risk of thromboembolic events, alongside higher costs [5,14,15].
- Protamine sulfate for unfractionated heparin (partial for LMWH).
Special Populations
- Neonates have lower vitamin K-dependent factors, which is why intramuscular vitamin K is given at birth to prevent hemorrhagic disease of the newborn.
- Pregnancy shifts the balance toward hypercoagulability (rising fibrinogen, factor VIII, vWF; falling protein S; reduced fibrinolysis). This protects against postpartum bleeding but increases VTE risk.
- Older adults often have higher D-dimer baselines and more comorbid bleeding risk factors, which complicates both diagnosis and anticoagulation.
Frequently Asked Questions (FAQs)
What is hemostasis in simple terms?
Hemostasis is the body's way of stopping bleeding after an injury and keeping blood flowing freely the rest of the time. It works in four overlapping stages: vessels narrow, platelets form a plug, clotting factors weave a fibrin mesh that stabilizes the plug, and finally an enzyme called plasmin dissolves the clot once healing is complete.
What is the difference between primary and secondary hemostasis?
Primary hemostasis is the immediate platelet response by adhesion to the vessel wall through von Willebrand factor and GPIb, activation, and aggregation through GPIIb/IIIa. Secondary hemostasis is the coagulation cascade, which generates thrombin and converts fibrinogen into a stable fibrin mesh that locks the platelet plug in place.
What are the main causes of bleeding disorders?
Bleeding disorders come from three sources: platelet problems (low count or impaired function), clotting factor deficiencies (such as hemophilia A and B, or von Willebrand disease), and vascular wall weakness. Acquired causes include liver disease, vitamin K deficiency, and anticoagulant medications.
Why do some people form clots when they shouldn't?
Thrombosis happens when the balance tips toward clotting. Risk factors include inherited thrombophilias (Factor V Leiden, prothrombin gene mutation, deficiencies of antithrombin, protein C, or protein S), acquired conditions (cancer, antiphospholipid syndrome), and situational triggers like surgery, immobility, or pregnancy. Virchow's triad summarizes the drivers: stasis, vessel injury, and a hypercoagulable state.
How are anticoagulants different from antiplatelet drugs?
Antiplatelets (aspirin, clopidogrel) block platelet activation and are used for arterial clots, where platelets dominate. Anticoagulants (heparin, warfarin, DOACs) block clotting factors and are used for venous clots, where fibrin dominates. Some patients receive both.
Can a clot or anticoagulant be reversed in an emergency?
Yes. Vitamin K and four-factor PCC reverse warfarin. Idarucizumab reverses dabigatran, and andexanet alfa reverses apixaban and rivaroxaban [5]. For active dangerous clots, thrombolytics like tPA can dissolve fibrin, but the bleeding risk is significant.
Glossary of Related Medical Terms
- ADAMTS13 — Plasma enzyme that trims very large vWF multimers; deficiency causes TTP.
- Coagulation cascade — Sequential activation of clotting factors leading to fibrin.
- D-dimer — Fibrin breakdown product used mainly to rule out clots.
- Endothelium — Inner lining of blood vessels; actively prevents clotting when intact.
- Fibrin / Fibrinogen — Insoluble clot protein and its soluble plasma precursor.
- Fibrinolysis — Plasmin-driven breakdown of fibrin once healing is complete.
- GPIb — Platelet receptor that binds vWF for adhesion.
- GPIIb/IIIa — Platelet receptor that binds fibrinogen for aggregation.
- Hemarthrosis — Bleeding into a joint, typical of severe hemophilia.
- Hemostasis — Coordinated process that stops bleeding while preserving blood flow.
- Petechiae — Pinpoint capillary bleeds, typical of low platelets.
- Plasmin — Active enzyme that dissolves fibrin.
- Platelet — Anucleate cell fragment from megakaryocytes; central to primary hemostasis.
- PT / aPTT — Tests of extrinsic and intrinsic coagulation pathways.
- Subendothelial matrix — Collagen and vWF layer beneath endothelium that triggers platelets when exposed.
- Thrombin — Central coagulation enzyme; converts fibrinogen to fibrin.
- Thrombocytopenia — Low platelet count.
- Thrombosis — Clot inside an intact vessel.
- Tissue factor (TF) — Membrane protein that initiates coagulation at injury sites.
- Vasoconstriction — Reflex narrowing of injured vessels.
- von Willebrand factor (vWF) — Plasma protein bridging platelets to collagen and stabilizing factor VIII.
Disclaimer: This article is intended for educational and informational purposes only. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- LaPelusa A, Dave HD. Physiology, Hemostasis. [Updated 2023 May 1]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2026 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK545263/
- Hoffman, M., & Monroe, D. M. (2001). A cell-based model of hemostasis. Thrombosis and Haemostasis, 85(6), 958–965. https://doi.org/10.1055/s-0037-1615947
- Smith, S. A., Travers, R. J., & Morrissey, J. H. (2015). How it all starts: Initiation of the clotting cascade. Critical reviews in biochemistry and molecular biology, 50(4), 326–336. https://doi.org/10.3109/10409238.2015.1050550
- Versteeg, H. H., Heemskerk, J. W., Levi, M., & Reitsma, P. H. (2013). New fundamentals in hemostasis. Physiological reviews, 93(1), 327–358. https://doi.org/10.1152/physrev.00016.2011
- Levy, J. H., Shaw, J. R., Castellucci, L. A., Connors, J. M., Douketis, J., Lindhoff-Last, E., Rocca, B., Samama, C. M., Siegal, D., & Weitz, J. I. (2024). Reversal of direct oral anticoagulants: guidance from the SSC of the ISTH. Journal of thrombosis and haemostasis : JTH, 22(10), 2889–2899. https://doi.org/10.1016/j.jtha.2024.07.009
- Mahlangu, J., Oldenburg, J., Paz-Priel, I., Negrier, C., Niggli, M., Mancuso, M. E., Schmitt, C., Jiménez-Yuste, V., Kempton, C., Dhalluin, C., Callaghan, M. U., Bujan, W., Shima, M., Adamkewicz, J. I., Asikanius, E., Levy, G. G., & Kruse-Jarres, R. (2018). Emicizumab Prophylaxis in Patients Who Have Hemophilia A without Inhibitors. The New England journal of medicine, 379(9), 811–822. https://doi.org/10.1056/NEJMoa1803550
- Ozelo, M. C., Mahlangu, J., Pasi, K. J., Giermasz, A., Leavitt, A. D., Laffan, M., Symington, E., Quon, D. V., Wang, J. D., Peerlinck, K., Pipe, S. W., Madan, B., Key, N. S., Pierce, G. F., O'Mahony, B., Kaczmarek, R., Henshaw, J., Lawal, A., Jayaram, K., Huang, M., … GENEr8-1 Trial Group (2022). Valoctocogene Roxaparvovec Gene Therapy for Hemophilia A. The New England journal of medicine, 386(11), 1013–1025. https://doi.org/10.1056/NEJMoa2113708
- Pipe, S. W., Leebeek, F. W. G., Recht, M., Key, N. S., Castaman, G., Miesbach, W., Lattimore, S., Peerlinck, K., Van der Valk, P., Coppens, M., Kampmann, P., Meijer, K., O'Connell, N., Pasi, K. J., Hart, D. P., Kazmi, R., Astermark, J., Hermans, C. R. J. R., Klamroth, R., Lemons, R., … Monahan, P. E. (2023). Gene Therapy with Etranacogene Dezaparvovec for Hemophilia B. The New England journal of medicine, 388(8), 706–718. https://doi.org/10.1056/NEJMoa2211644
- Scully, M., Cataland, S. R., Peyvandi, F., Coppo, P., Knöbl, P., Kremer Hovinga, J. A., Metjian, A., de la Rubia, J., Pavenski, K., Callewaert, F., Biswas, D., De Winter, H., Zeldin, R. K., & HERCULES Investigators (2019). Caplacizumab Treatment for Acquired Thrombotic Thrombocytopenic Purpura. The New England journal of medicine, 380(4), 335–346. https://doi.org/10.1056/NEJMoa1806311
- Furie, B., & Furie, B. C. (2008). Mechanisms of thrombus formation. The New England journal of medicine, 359(9), 938–949. https://doi.org/10.1056/NEJMra0801082
- Young, G., Srivastava, A., Kavakli, K., Ross, C., Sathar, J., You, C. W., Tran, H., Sun, J., Wu, R., Poloskey, S., Qiu, Z., Kichou, S., Andersson, S., Mei, B., & Rangarajan, S. (2023). Efficacy and safety of fitusiran prophylaxis in people with haemophilia A or haemophilia B with inhibitors (ATLAS-INH): a multicentre, open-label, randomised phase 3 trial. Lancet (London, England), 401(10386), 1427–1437. https://doi.org/10.1016/S0140-6736(23)00284-2
- Matino, D., Palladino, A., Taylor, C. T., Hwang, E., Raje, S., Nayak, S., McDonald, R., Acharya, S. S., Mahlangu, J., Jiménez-Yuste, V., Choraria, N., Yang, R., Li, C. K., Al-Khabori, M., Wali, Y., Morales Adrián, J., Park, Y. S., Zülfikar, O. B., & Teeter, J. (2025). Marstacimab prophylaxis in hemophilia A/B without inhibitors: results from the phase 3 BASIS trial. Blood, 146(14), 1654–1663. https://doi.org/10.1182/blood.2024027468
- Miesbach W. (2026). Gene Therapy of Haemophilia: Current Status and Future Directions. Hamostaseologie, 46(1), 10–16. https://doi.org/10.1055/a-2751-7625
- Orso, D., Fonda, F., Brussa, A., Comisso, I., Auci, E., Sartori, M., & Bove, T. (2024). Andexanet alpha versus four-factor prothrombin complex concentrate in DOACs anticoagulation reversal: an updated systematic review and meta-analysis. Critical care (London, England), 28(1), 221. https://doi.org/10.1186/s13054-024-05014-x
- Koo, S. J., Hussain, Y., Booth, D. Y., Desai, P., Oh, E. S., Rios, J., & Audley, K. (2024). Four-factor prothrombin complex concentrate versus andexanet alfa for direct oral anticoagulant reversal. Journal of the American Pharmacists Association : JAPhA, 64(2), 395–401. https://doi.org/10.1016/j.japh.2023.11.015