Key Takeaways
The coagulation cascade is the second stage of hemostasis. Once platelets form their initial plug, clotting factors weave a fibrin mesh that locks the plug in place and seals the injury.
- Two pathways ▾: Two pathways feed into a common pathway. The extrinsic pathway begins with tissue factor outside the vessel; the intrinsic pathway is triggered by contact in the test tube. They converge on Factor X, which leads to a burst of thrombin and a fibrin clot [1,12].
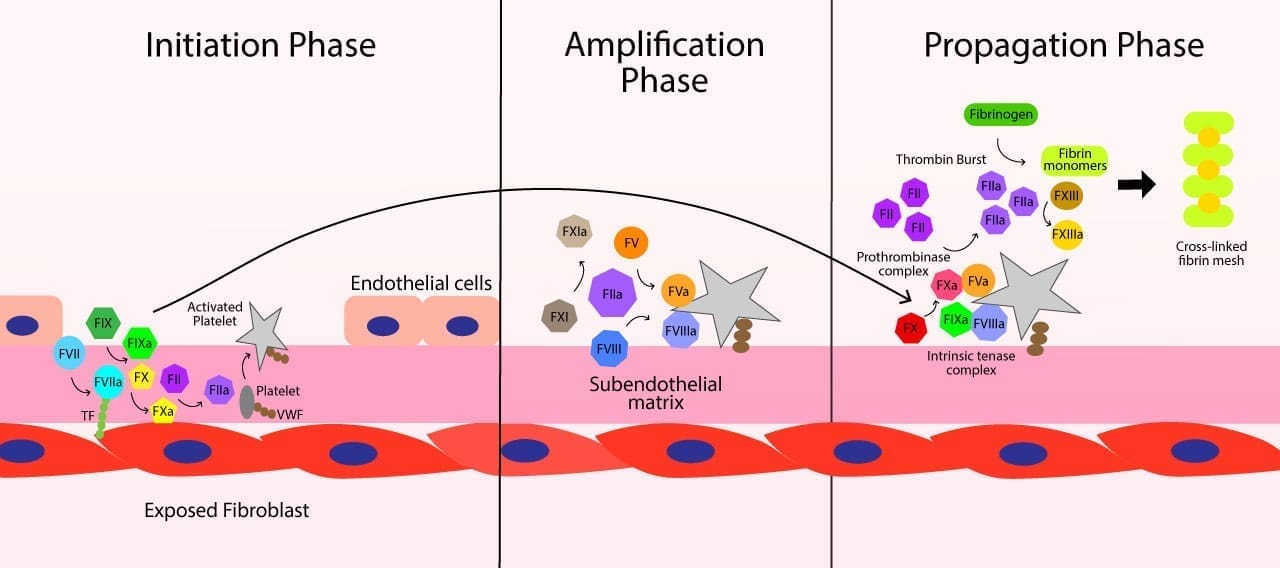
- Cell-based model ▾: In the body, coagulation actually unfolds in three overlapping phases on cell surfaces: initiation, amplification, and propagation. This is the cell-based model of Hoffman and Monroe [1,2,4].
- Investigations ▾: Two screening tests cover most of the cascade. PT/INR reflects the extrinsic and common pathways and monitors warfarin. APTT reflects the intrinsic and common pathways and monitors unfractionated heparin [12].
- Related bleeding disorders ▾: Disorders fall into two groups. Bleeding disorders (such as hemophilia A and B) reflect insufficient clotting; thrombotic disorders (such as Factor V Leiden) reflect excessive clotting.
- Treatment and management ▾: Modern pharmacology targets specific points in the coagulation cascade. Direct oral anticoagulants block thrombin or Factor Xa, and Factor XI inhibitors are emerging as a potentially safer next class [6,7].
*Click ▾ for more information
Introduction
Bleeding is dangerous. So is clotting in the wrong place. Your body has to do both — stop blood loss after injury, and keep blood flowing everywhere else. That balance is the job of hemostasis. This article focuses on the second half of the process: the coagulation cascade, also called secondary hemostasis.
The Two Phases of Hemostasis
Hemostasis runs in two phases that work seamlessly together.
- Primary hemostasis is the immediate response. Within seconds of injury, platelets stick to exposed collagen, become activated, and clump into a temporary plug.
- Secondary hemostasis or the coagulation cascade is the reinforcement. Over the next few minutes, plasma clotting factors generate thrombin, which converts fibrinogen into a tough fibrin mesh. The mesh stabilizes the platelet plug into a durable clot.
The system is also self-limiting. Natural anticoagulants such as antithrombin and Protein C constantly check clotting so it stays at the injury site and does not spread.
Coagulation Factors: The Players
Most coagulation factors fall into one of these categories:
- Zymogens — inactive protein precursors waiting to be cut into their active enzyme form. Examples: prothrombin, Factor X, Factor IX.
- Serine proteases — the activated enzymes (the lowercase "a" in Xa, IXa, IIa). They cut their next target in line.
- Cofactors — non-enzymatic helpers that dramatically speed up reactions. Factors V and VIII are the key cofactors.
- Carrier proteins — von Willebrand factor (VWF) is primarily known for mediating platelet adhesion, but it also physically binds to Factor VIII in the plasma. This binding protects Factor VIII from premature degradation by Protein C. Without VWF, Factor VIII levels drop drastically, which is why severe von Willebrand disease can mimic the secondary hemostasis failure seen in hemophilia A [14].
- Calcium ions (Ca²⁺) — needed at almost every step. Calcium binds to specific carboxylated regions of vitamin K–dependent factors and anchors them to phospholipid surfaces.
Why the Numbers Look Random
Coagulation factors are numbered in the order they were discovered, not in the order they act. Roman numerals are historical convention. A lowercase "a" indicates the activated form (Factor Xa is activated Factor X). Some factors have alternative names for example Factor VIII is "antihemophilic factor," Factor IX is "Christmas factor" (after the patient).
| Factor | How It Gets Activated | What It Does |
|---|---|---|
| Tissue Factor (TF, III) | Exposed at injury | Starts the extrinsic pathway |
| Factor VII | Binds TF, with calcium | Activates Factors X and IX |
| Factor X | Cleaved by VIIa or IXa-VIIIa, with calcium | Forms prothrombinase |
| Factor IX | Cleaved by VIIa or XIa | Joins VIIIa to form tenase |
| Factor VIII | Cleaved by thrombin | Cofactor for IXa |
| Prothrombin (II) | Cleaved by Xa-Va | Becomes thrombin |
| Thrombin (IIa) | Made by prothrombinase | Cleaves fibrinogen, activates V, VIII, XI, XIII |
| Fibrinogen (I) | Cleaved by thrombin | Becomes fibrin monomers |
| Factor XIII | Activated by thrombin | Cross-links fibrin into a stable mesh |
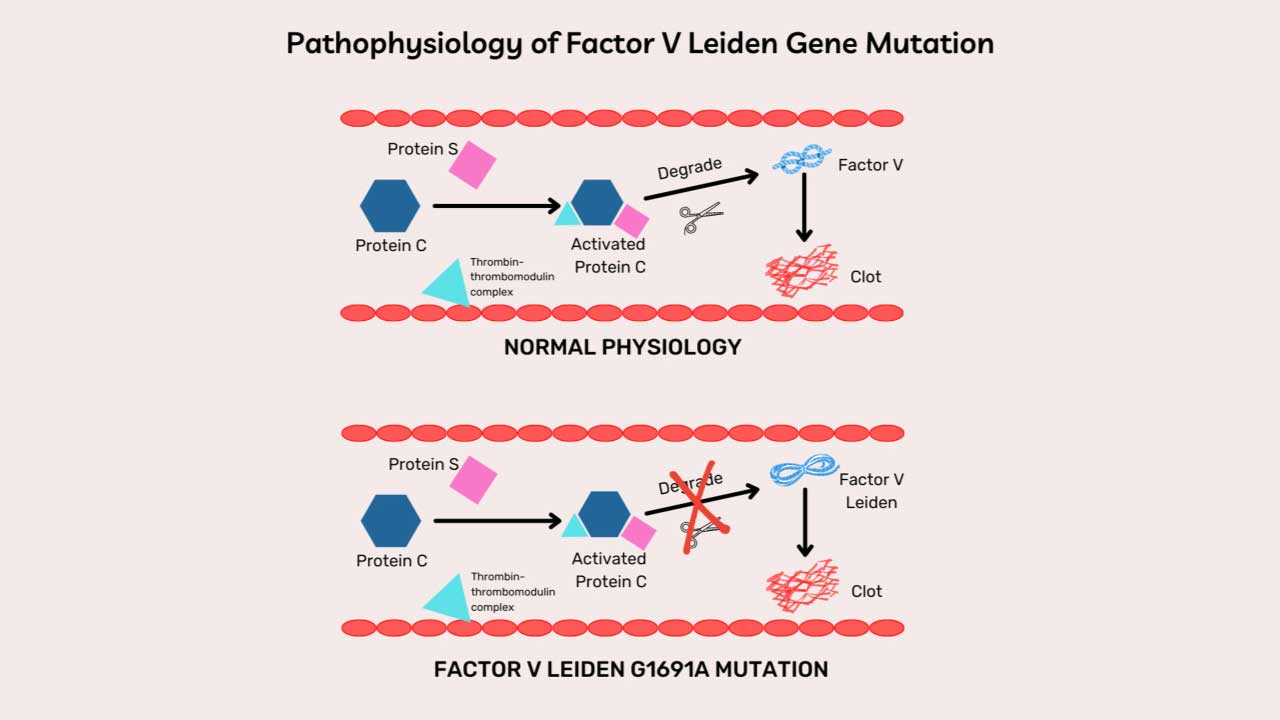
| Protein C | Activated by thrombin–thrombomodulin | Inactivates Va and VIIIa |
The Coagulation Cascade
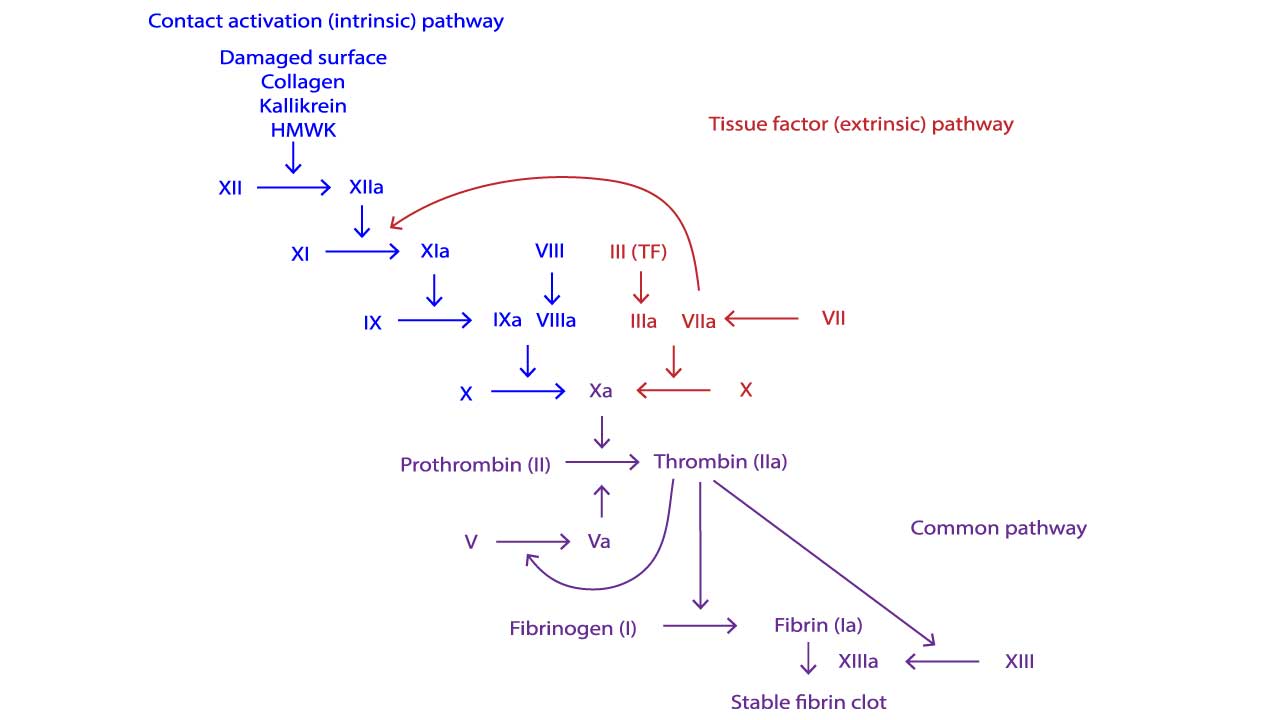
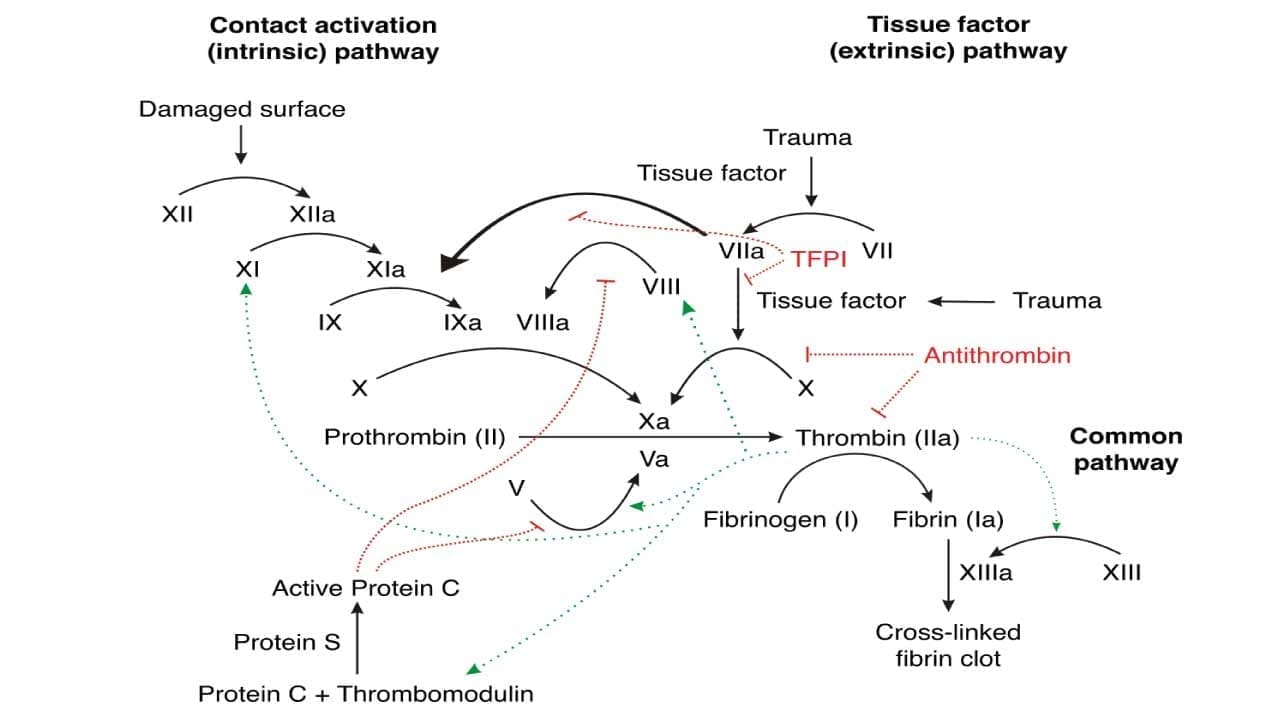
Extrinsic Pathway
This is what fires first in vivo. Injury exposes tissue factor (TF) on subendothelial cells to circulating Factor VII. TF binds and activates Factor VII to VIIa. The TF–VIIa complex then activates Factor X to Xa and a small amount of Factor IX to IXa. Factor Xa, with its cofactor Factor Va, converts a small amount of prothrombin to thrombin. PT/INR is the laboratory test that maps onto this pathway [12].
Intrinsic Pathway
The intrinsic pathway is best understood as the test-tube pathway. In a coagulation analyzer, contact between plasma and a negatively charged activator (silica, kaolin) activates Factor XII to XIIa. XIIa activates XI to XIa, XIa activates IX to IXa, and IXa (together with cofactor VIIIa and calcium on a phospholipid surface) activates X to Xa. APTT measures this pathway. In the body, a separate route to IXa exists: thrombin itself activates Factor XI as part of the amplification phase (described below).
Common Pathway
Both pathways converge at Factor Xa. Factor Xa plus Factor Va on a phospholipid surface forms the prothrombinase complex, which converts prothrombin (II) to thrombin (IIa).
Thrombin is the central engine of clotting. It cleaves fibrinogen into fibrin monomers, activates Factor XIII to cross-link them, and feeds back to activate Factors V, VIII, and XI to amplify itself. Activated platelets supply the phospholipid surface and additional Factor V, fibrinogen, and Factor XIII from their α-granules [9,13].
Fibrin Formation and Clot Stabilization
The end goal is a stable fibrin mesh.
From fibrinogen to fibrin monomers. Thrombin cleaves fibrinogen at specific sites, releasing fibrinopeptides A and B. The trimmed monomers expose binding regions and self-assemble into long protofibrils.
Cross-linking. Activated Factor XIII (Factor XIIIa) is a transglutaminase. It forms covalent bonds between adjacent fibrin strands, turning a loose mesh into a tough, insoluble lattice that resists breakdown.
Calcium's role. Calcium does not directly cleave anything in this step, but it stabilizes thrombin and Factor XIIIa and supports their interactions with fibrin. Without it, the mesh forms slowly or not at all.
The cross-linked fibrin mesh does three things at once: it strengthens the clot, restricts clotting to the injury site, and provides scaffolding for cells migrating in to repair the vessel.
The Cell-based Model
The traditional waterfall cascade has served generations of students, but it has a problem. It cannot explain why patients with Factor XII deficiency do not bleed despite a markedly prolonged APTT, or why hemophilia A causes such severe bleeding even though Factor VIII is "just" a cofactor.
In 2001, Hoffman and Monroe proposed the cell-based model of coagulation [1,2]. It is now the standard physiologic framework. The cascade is not wrong as it is the right way to understand what PT and APTT measure in vitro. But in vivo, clotting happens on cells, in three overlapping phases.
Phase 1: Initiation
Vessel injury exposes tissue factor on subendothelial cells (typically fibroblasts). Circulating Factor VII binds and is activated. The TF–VIIa complex activates small amounts of Factor X and Factor IX. The little Xa generated, with Factor Va, makes a small amount of thrombin. This first wave is too small to form a clot. Its purpose is to prime the next phase.
Phase 2: Amplification
That small amount of thrombin is the spark. It does several things at once:
- Activates platelets, exposing a negatively charged phospholipid surface (phosphatidylserine).
- Activates cofactors V and VIII on the platelet surface.
- Activates Factor XI to XIa on the platelet surface. This direct, thrombin-mediated activation of Factor XI is the crucial step that completely bypasses Factor XII. It explains the physiological mystery of why patients with severe Factor XII deficiency exhibit a prolonged APTT in the laboratory but do not experience clinical bleeding in vivo [1,15].
Now the stage is set.
Phase 3: Propagation
On the activated platelet surface, Factor IXa (made earlier and on the spot) binds Factor VIIIa to form the intrinsic tenase complex, which efficiently activates large amounts of Factor X. Factor Xa joins Factor Va to form the prothrombinase complex, which produces a thrombin burst (the burst that actually makes a useful clot). That thrombin cleaves fibrinogen and activates Factor XIII. The result is a cross-linked fibrin mesh, anchored to platelets, at the precise site of injury [1,2,3].
Why two models?
Because each answers a different question.
- The cascade model explains laboratory clotting tests. PT measures the extrinsic and common pathways. APTT measures the intrinsic and common pathways.
- The cell-based model explains in vivo hemostasis. It clarifies why platelets and cell surfaces matter, why Factor XII deficiency is asymptomatic (the in vivo system bypasses XII), and why hemophilia A and B are severe (Factor VIIIa and IXa are essential for the propagation thrombin burst) [4].
A 2023 Blood review by Toh and Yong argues for a convergent model that further integrates innate immune activation with coagulation, reflecting the increasing importance of inflammation–thrombosis crosstalk [4].
Comparison of The Traditional Coagulation Cascade vs Cell-Based Models
| Feature | Traditional Cascade | Cell-Based Model |
|---|---|---|
| Where it happens | Plasma | On cell surfaces |
| Initiator | Factor XII (intrinsic) or TF (extrinsic) | TF-bearing cells |
| Role of platelets | Passive surface | Active platform for amplification and propagation |
| Best at explaining | PT and APTT lab results | In vivo hemostasis and bleeding phenotypes |
Coagulation Inhibitors
Clotting must stop where the injury ends. Several systems make sure of that.
- Tissue factor pathway inhibitor (TFPI). Made in endothelial cells, TFPI binds Factor Xa and then docks onto the TF–VIIa complex, shutting down the extrinsic pathway after only a small amount of Xa is made. This forces the system to rely on the amplification loop for sustained clotting [9].
- Antithrombin (AT). The most important plasma inhibitor. It inactivates thrombin and Factors IXa, Xa, XIa, and XIIa. Heparin accelerates this binding by 1,000-fold or more, which is its mechanism of action.
- Protein C and Protein S. Thrombin bound to thrombomodulin on healthy endothelium activates Protein C. Protein C, with cofactor Protein S, inactivates Factors Va and VIIIa, dampening the cascade. Both proteins are vitamin K–dependent.
- Thrombomodulin. Beyond activating Protein C, the thrombin–thrombomodulin complex activates TAFI (thrombin-activatable fibrinolysis inhibitor), which links coagulation control to clot breakdown.
Dysfunction of the Coagulation Cascade
Cascade dysfunction goes one of two ways.
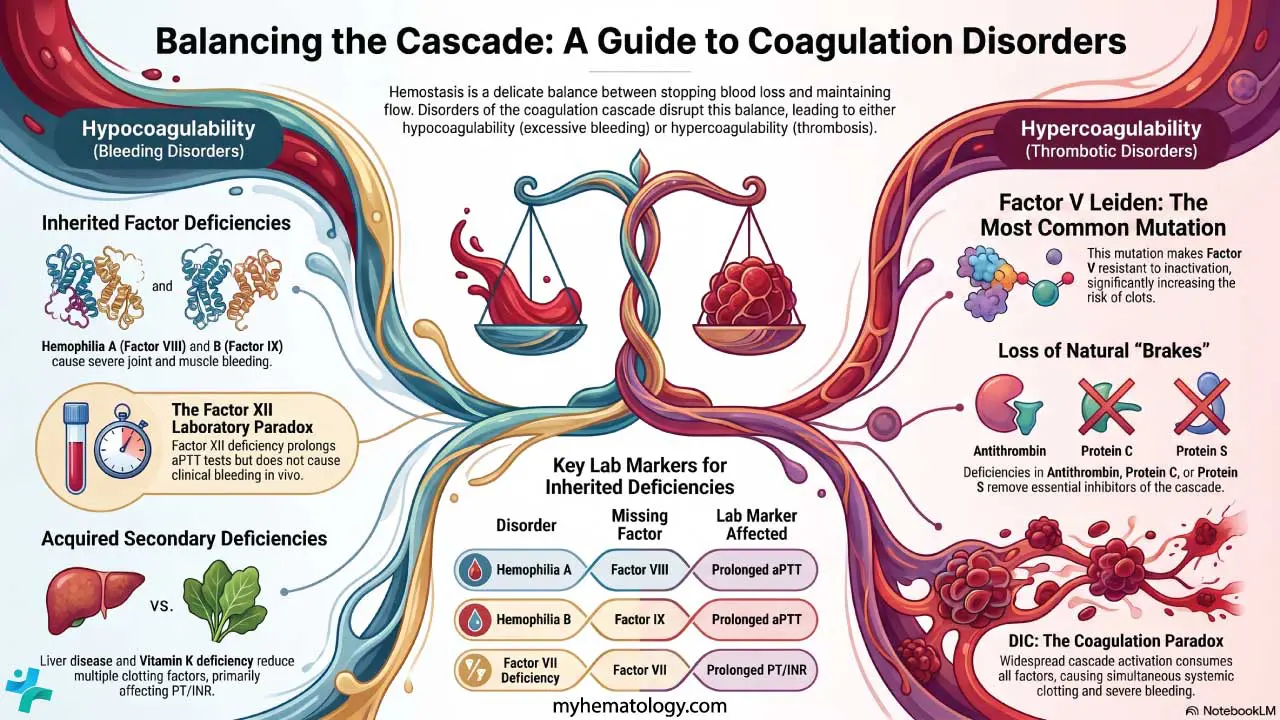
Bleeding Disorders (Hypocoagulability)
Inherited factor deficiencies
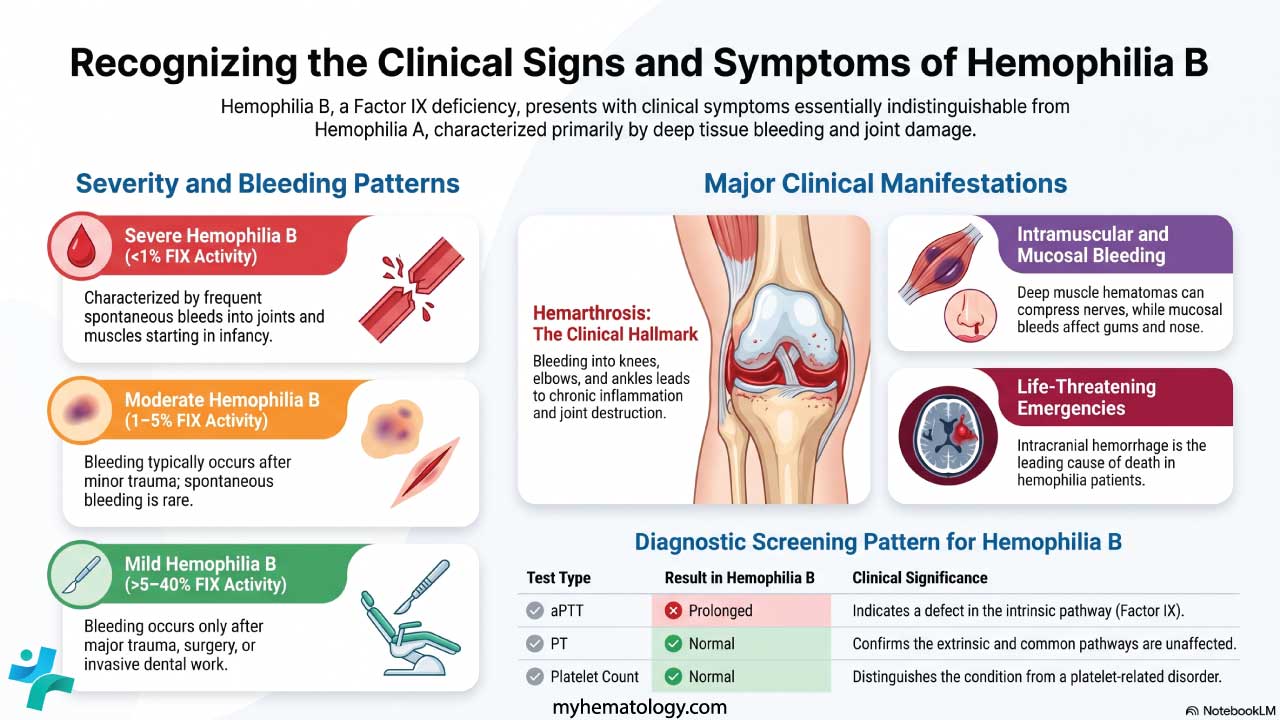
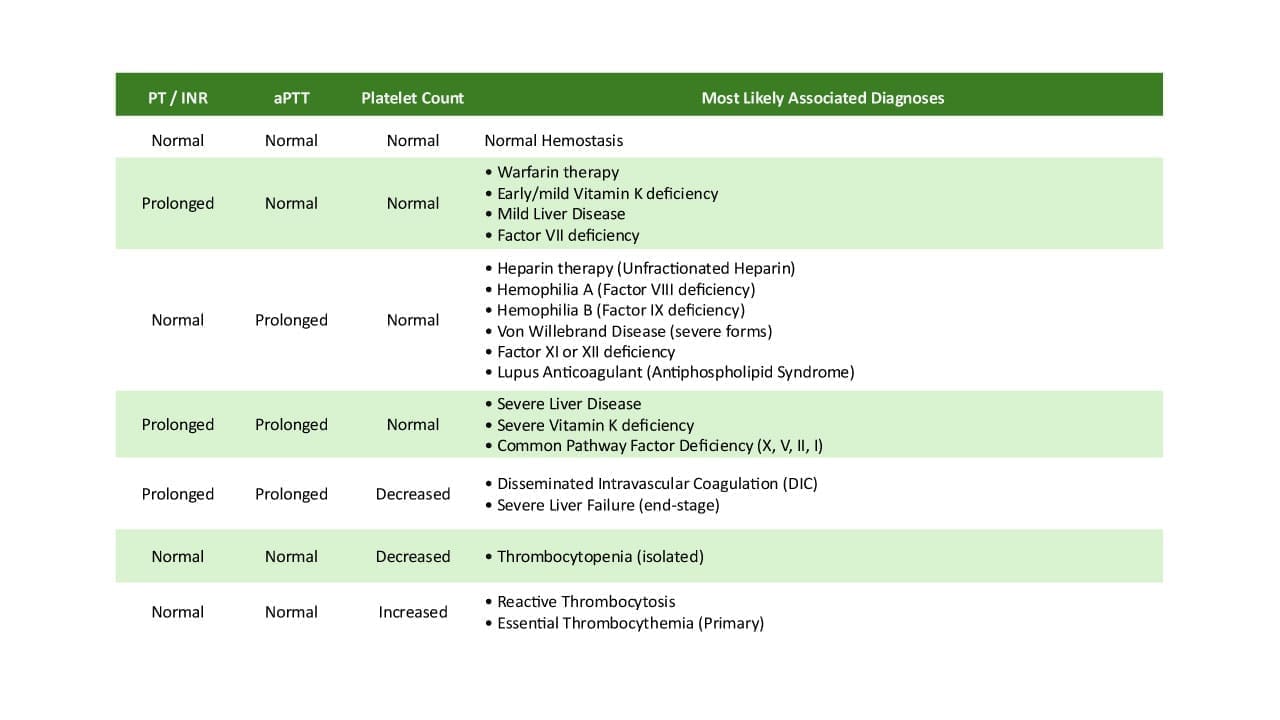
- Hemophilia A — Factor VIII deficiency. Severe bleeding into joints and muscles. APTT prolonged, PT normal.
- Hemophilia B — Factor IX deficiency. Same clinical picture as hemophilia A.
- Factor VII deficiency — affects the extrinsic pathway. Prolonged PT.
- Factor X, XI, or XIII deficiency — variable bleeding tendency.
- Congenital afibrinogenemia — absence of fibrinogen.
- Factor XII deficiency — prolonged APTT but no bleeding, because in vivo coagulation does not depend on XII. A textbook example of why the cell-based model matters.
Acquired deficiencies
- Liver disease — the liver makes most coagulation factors. In advanced liver disease, multiple factors fall together.
- Vitamin K deficiency — vitamin K is required for Factors II, VII, IX, and X plus Proteins C and S. Factor VII goes first because it has the shortest half-life, so PT prolongs before APTT.
- Disseminated intravascular coagulation (DIC) — widespread activation of coagulation consumes platelets and factors, producing simultaneous clotting and bleeding.
Coagulation Cascade Evaluation
Two tests do most of the heavy lifting.
- Prothrombin Time (PT) and INR — extrinsic and common pathways. Sensitive to Factors VII, X, V, II, and I. Used to monitor warfarin. Normal PT is roughly 11–15 seconds; INR around 0.9–1.3 [12].
- Activated Partial Thromboplastin Time (APTT) — intrinsic and common pathways. Sensitive to Factors XII, XI, IX, VIII, X, V, II, and I. Used to monitor unfractionated heparin. Normal APTT is roughly 25–40 seconds.
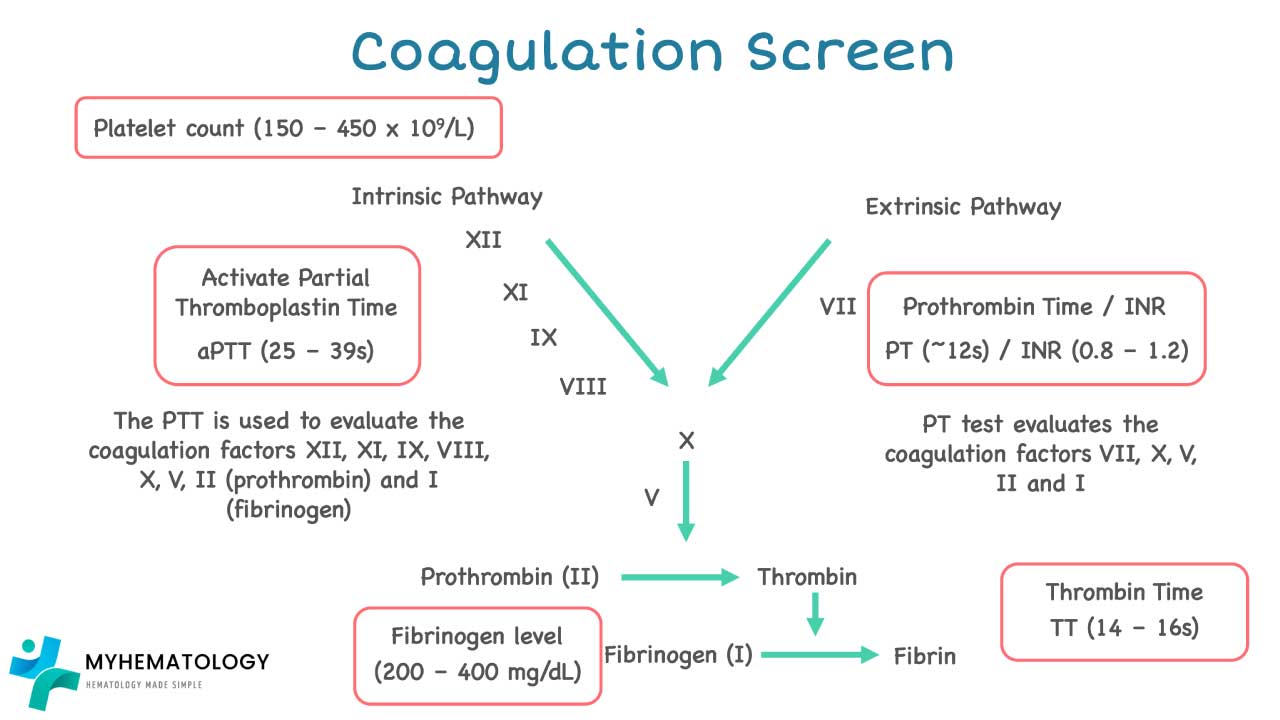
Other useful tests:
- Thrombin time (TT) — measures fibrinogen-to-fibrin conversion. Prolonged by heparin, low or abnormal fibrinogen, and direct thrombin inhibitors.
- Fibrinogen level — falls in DIC and severe liver disease; rises as an acute phase reactant.
- Activated clotting time (ACT) — point-of-care, used to monitor high-dose heparin during cardiac bypass and ECMO.
- Mixing studies. When PT or APTT is prolonged, mix patient plasma 1:1 with normal plasma and retest. Correction suggests factor deficiency. No correction suggests an inhibitor (heparin, lupus anticoagulant, or a specific factor antibody).
- Specific factor assays — measure individual clotting factor activity to diagnose hemophilia and other factor deficiencies.
- Viscoelastic tests (TEG, ROTEM) — assess whole-blood clotting in real time, including platelet–fibrin interaction. Increasingly used in trauma, cardiac surgery, and obstetric hemorrhage.
Thrombotic Disorders (Hypercoagulability)
Inherited thrombophilias
- Factor V Leiden. A mutation in Factor V that resists inactivation by activated Protein C. The most common inherited thrombophilia.
- Prothrombin G20210A. A mutation that raises prothrombin levels, increasing thrombin generation.
Anticoagulant protein deficiencies
- Antithrombin deficiency.
- Protein C and Protein S deficiency.
- TFPI deficiency (rare).
Each removes a brake from the coagulation cascade and tilts the balance toward clotting.
Treatment and Management
Once you know which step in the coagulation cascade is failing, you can target it directly.
Procoagulants — replacing what is missing
- Factor concentrates. Recombinant or plasma-derived Factor VIII for hemophilia A, Factor IX for hemophilia B.
- Prothrombin complex concentrates (PCCs). Contain Factors II, VII, IX, X (and sometimes Proteins C and S). First-line for urgent reversal of warfarin.
- Cryoprecipitate. Rich in fibrinogen, Factor VIII, von Willebrand factor, and Factor XIII. Used in hypofibrinogenemia.
- Fresh frozen plasma (FFP). Contains all clotting factors. Used in liver disease, DIC, and massive transfusion protocols.
- Recombinant activated Factor VII (rFVIIa). Bypasses the intrinsic and extrinsic pathways. Used in patients with inhibitors against Factor VIII or IX.
- Desmopressin (DDAVP). Releases stored von Willebrand factor and Factor VIII. Useful in mild hemophilia A and certain types of von Willebrand disease.
- Vitamin K. Reverses warfarin and treats vitamin K deficiency.
Newer hemophilia therapies
The hemophilia treatment landscape has changed substantially in the past decade [10,11].
- Emicizumab — a bispecific antibody that mimics Factor VIIIa by bridging Factors IXa and X. Standard prophylaxis in hemophilia A with or without inhibitors.
- Efanesoctocog alfa — FDA approved in 2023. A high-sustained Factor VIII replacement that overcomes the von Willebrand factor half-life ceiling, dosed once weekly [10].
- Anti-TFPI agents — concizumab (approved in several jurisdictions) and marstacimab (FDA approved October 2024) rebalance hemostasis by blocking TFPI.
- RNA interference (RNAi) therapies — fitusiran is a subcutaneously administered small interfering RNA (siRNA) that targets and lowers antithrombin production in the liver. By deliberately silencing this natural "brake" on the coagulation cascade, fitusiran restores adequate thrombin generation in patients with hemophilia A or B, regardless of whether they have developed inhibitors [16].
- AAV gene therapy — etranacogene dezaparvovec for hemophilia B and valoctocogene roxaparvovec for hemophilia A are FDA approved.
Anticoagulants — slowing the coagulation cascade
- Heparin (unfractionated and LMWH) and fondaparinux. Boost antithrombin. Unfractionated heparin is monitored by APTT; high-dose heparin during cardiac surgery is monitored by ACT. LMWH and fondaparinux are usually given at fixed weight-based doses.
- Warfarin. Blocks vitamin K recycling, lowering active Factors II, VII, IX, X plus Proteins C and S. Monitored by PT/INR. A teaching point: because Protein C has a short half-life, warfarin can briefly tilt patients toward clotting at initiation. Heparin overlap is the standard workaround.
- Direct oral anticoagulants (DOACs).
- Direct thrombin inhibitors — dabigatran (oral), argatroban and bivalirudin (IV).
- Direct Factor Xa inhibitors — apixaban, rivaroxaban, edoxaban. Widely used for atrial fibrillation, DVT, and PE; routine monitoring is not required.

Reversal of Anticoagulants
DOACs were once seen as a weakness of the system because they could not be reversed quickly. That has changed [6,7].
- Idarucizumab — a monoclonal antibody fragment that binds dabigatran with very high affinity. Used in major bleeding or before urgent surgery.
- Andexanet alfa — a recombinant decoy of Factor Xa. Sequesters apixaban and rivaroxaban. The 2024 ANNEXA-I trial showed it improved hemostasis in factor Xa inhibitor–associated intracerebral hemorrhage compared with usual care, though with a higher rate of thrombotic events [7].
- PCCs (Kcentra) — used off-label for factor Xa inhibitor reversal where andexanet alfa is unavailable; first-line for urgent warfarin reversal.
Factor XI Inhibitors: The Next Generation
People with congenital Factor XI deficiency have only mild bleeding, even though they protect against thrombosis. That observation has driven a new drug class.
- Abelacimab — a monoclonal antibody against Factor XI. The phase 2 AZALEA-TIMI 71 trial in atrial fibrillation was stopped early because bleeding was substantially lower than with rivaroxaban.
- Asundexian — a small-molecule Factor XIa inhibitor. The phase 3 OCEANIC-AF trial in atrial fibrillation was stopped early for inferior efficacy versus apixaban.
- Milvexian — also a Factor XIa inhibitor, in ongoing phase 3 trials.
The class is not yet ready for routine practice, but the rationale of separating thrombosis prevention from bleeding risk is important to understand because it follows directly from the cell-based model of the coagulation cascade. As this drug class evolves, clinical focus has increasingly shifted toward specific high-risk populations, particularly patients with end-stage kidney disease (ESKD) on hemodialysis. In these patients, traditional DOACs often carry prohibitively high bleeding risks, making the uncoupling of hemostasis and thrombosis offered by Factor XI inhibitors highly desirable [17,18].
Topical and adjunctive agents
Topical thrombin, fibrin glue, and hemostatic powders such as zeolites or TC-325 promote local clotting in surgery and trauma. They concentrate clotting factors at a bleeding site without affecting systemic coagulation.
Frequently Asked Questions (FAQs)
What is the coagulation cascade in simple terms?
The coagulation cascade is the chain of protein reactions in your blood that builds a clot when a vessel is injured. Inactive proteins called clotting factors are switched on one after another, ending in a fibrin mesh that holds platelets together and seals the wound. The system has built-in brakes so clotting stays at the injury site and does not spread.
What is the difference between the intrinsic and extrinsic pathways?
The names come from the laboratory, not the body. The extrinsic pathway needs a substance from outside the blood (tissue factor) to start, and is measured by the PT/INR test. The intrinsic pathway can be triggered by something already in the blood (a charged surface in the test tube) and is measured by the APTT. Inside the body, both pathways merge into a single cell-based process that begins on tissue factor–bearing cells and accelerates on platelets.
Why does warfarin take days to work?
Warfarin blocks vitamin K, which is needed to finish making Factors II, VII, IX, and X, plus the natural anticoagulants Protein C and Protein S. The clotting factors already in the blood must be cleared first, and they have different half-lives. Factor VII goes first, but Factor II (prothrombin) lingers for days. Until prothrombin levels drop, warfarin is not fully effective. Protein C also drops quickly, which is why patients can briefly tilt toward more clotting just after starting warfarin but usually managed with overlapping heparin.
Why do hemophilia A and B prolong APTT but not PT?
Hemophilia A is Factor VIII deficiency and hemophilia B is Factor IX deficiency. Both factors sit in the intrinsic pathway, which is what APTT measures, so APTT is prolonged. The extrinsic pathway (Factor VII and tissue factor) is unaffected, so PT stays normal. Bleeding tendency does not depend on which test is abnormal but it depends on which factor is missing.
Are there newer anticoagulants beyond warfarin and heparin?
Yes. Direct oral anticoagulants, or DOACs, are now first-line for most non-valvular atrial fibrillation and for treating deep vein thrombosis and pulmonary embolism. Dabigatran blocks thrombin directly. Apixaban, rivaroxaban, and edoxaban block Factor Xa directly. They are easier to dose than warfarin and do not need routine monitoring. Specific reversal agents now exist: idarucizumab for dabigatran and andexanet alfa for the Factor Xa inhibitors. A newer class for example Factor XI inhibitors such as abelacimab, milvexian, and asundexian, is in late-stage trials and aims to prevent clots with less bleeding.
What is the cell-based model and why does it matter?
The cell-based model, proposed by Hoffman and Monroe in 2001, replaced the older "waterfall" cascade as the way coagulation actually works in the body. It splits clotting into three overlapping phases — initiation on tissue factor–bearing cells, amplification on activating platelets, and propagation as a thrombin burst on the platelet surface. The cascade diagram is still useful for understanding lab tests like PT and APTT, but the cell-based model better explains why some factor deficiencies cause severe bleeding (Factor VIII, IX) while others do not (Factor XII).
Glossary of Related Medical Term
- Coagulation cascade: the series of protein-cutting reactions in plasma that ends in a fibrin clot. Often shown as intrinsic, extrinsic, and common pathways.
- Hemostasis: the body's process of stopping bleeding from a damaged vessel without blocking healthy blood flow.
- Primary hemostasis: the first response, where platelets stick to the injured wall and clump together to form a temporary plug.
- Secondary hemostasis: the reinforcement step, where coagulation factors weave a fibrin mesh that locks the platelet plug in place.
- Zymogen: an inactive form of a protein-cutting enzyme that becomes active only when needed.
- Serine protease: an enzyme (such as thrombin or Factor Xa) that activates other clotting factors by cutting them at specific sites.
- Cofactor: a partner protein (such as Factor V or Factor VIII) that is not enzymatic but speeds up the activity of a serine protease.
- Tissue factor (TF, Factor III): a protein on cells outside the blood vessel; exposed by injury, it triggers the extrinsic pathway.
- Thrombin (Factor IIa): the central clotting enzyme; it converts fibrinogen to fibrin and activates other factors.
- Fibrinogen (Factor I): a soluble plasma protein turned into the fibrin mesh by thrombin.
- Fibrin: the insoluble protein that forms the meshwork of a blood clot.
- Tenase complex: Factor IXa plus Factor VIIIa on a platelet surface; activates Factor X.
- Prothrombinase complex: Factor Xa plus Factor Va on a platelet surface; converts prothrombin to thrombin.
- Antithrombin (AT): a natural anticoagulant in plasma that inactivates thrombin and Factor Xa, especially when bound to heparin.
- Protein C / Protein S: vitamin K–dependent natural anticoagulants that switch off Factors Va and VIIIa.
- Tissue factor pathway inhibitor (TFPI): shuts down the early extrinsic pathway after a small amount of thrombin has been made.
- Hemophilia A / B: inherited bleeding disorders caused by deficiency of Factor VIII (A) or Factor IX (B).
- DOACs: direct oral anticoagulants — drugs that block thrombin (dabigatran) or Factor Xa (apixaban, rivaroxaban, edoxaban).
- PT / INR: prothrombin time (and its standardized ratio) — assesses the extrinsic and common pathways; monitors warfarin.
- APTT (aPTT): activated partial thromboplastin time — assesses the intrinsic and common pathways; monitors heparin.
- Thrombophilia: a tendency to clot inappropriately, inherited or acquired.
Disclaimer: This article is intended for educational and informational purposes only . It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Hoffman, M., & Monroe, D. M., 3rd (2001). A cell-based model of hemostasis. Thrombosis and haemostasis, 85(6), 958–965.
- Hoffman, M., & Monroe, D. M. (2007). Coagulation 2006: a modern view of hemostasis. Hematology/oncology clinics of North America, 21(1), 1–11. https://doi.org/10.1016/j.hoc.2006.11.004
- Smith S. A. (2009). The cell-based model of coagulation. Journal of veterinary emergency and critical care (San Antonio, Tex. : 2001), 19(1), 3–10. https://doi.org/10.1111/j.1476-4431.2009.00389.x
- Yong, J., & Toh, C. H. (2023). Rethinking coagulation: from enzymatic cascade and cell-based reactions to a convergent model involving innate immune activation. Blood, 142(25), 2133–2145. https://doi.org/10.1182/blood.2023021166
- Troisi, R., Balasco, N., Autiero, I., Sica, F., & Vitagliano, L. (2023). New insight into the traditional model of the coagulation cascade and its regulation: illustrated review of a three-dimensional view. Research and practice in thrombosis and haemostasis, 7(6), 102160. https://doi.org/10.1016/j.rpth.2023.102160
- Levy, J. H., Shaw, J. R., Castellucci, L. A., Connors, J. M., Douketis, J., Lindhoff-Last, E., Rocca, B., Samama, C. M., Siegal, D., & Weitz, J. I. (2024). Reversal of direct oral anticoagulants: guidance from the SSC of the ISTH. Journal of thrombosis and haemostasis : JTH, 22(10), 2889–2899. https://doi.org/10.1016/j.jtha.2024.07.009
- Connolly, S. J., Sharma, M., Cohen, A. T., Demchuk, A. M., Członkowska, A., Lindgren, A. G., Molina, C. A., Bereczki, D., Toni, D., Seiffge, D. J., Tanne, D., Sandset, E. C., Tsivgoulis, G., Christensen, H., Beyer-Westendorf, J., Coutinho, J. M., Crowther, M., Verhamme, P., Amarenco, P., Roine, R. O., … ANNEXA-I Investigators (2024). Andexanet for Factor Xa Inhibitor-Associated Acute Intracerebral Hemorrhage. The New England journal of medicine, 390(19), 1745–1755. https://doi.org/10.1056/NEJMoa2313040
- Park, S., & Park, J. K. (2024). Back to basics: the coagulation pathway. Blood research, 59(1), 35. https://doi.org/10.1007/s44313-024-00040-8
- Al-Koussa, H., AlZaim, I., & El-Sabban, M. E. (2022). Pathophysiology of Coagulation and Emerging Roles for Extracellular Vesicles in Coagulation Cascades and Disorders. Journal of clinical medicine, 11(16), 4932. https://doi.org/10.3390/jcm11164932
- Konkle, B. A., Shapiro, A. D., Quon, D. V., Staber, J. M., Kulkarni, R., Ragni, M. V., Chhabra, E. S., Poloskey, S., Rice, K., Katragadda, S., Fruebis, J., & Benson, C. C. (2020). BIVV001 Fusion Protein as Factor VIII Replacement Therapy for Hemophilia A. The New England journal of medicine, 383(11), 1018–1027. https://doi.org/10.1056/NEJMoa2002699
- Mahlangu, J., Oldenburg, J., Paz-Priel, I., Negrier, C., Niggli, M., Mancuso, M. E., Schmitt, C., Jiménez-Yuste, V., Kempton, C., Dhalluin, C., Callaghan, M. U., Bujan, W., Shima, M., Adamkewicz, J. I., Asikanius, E., Levy, G. G., & Kruse-Jarres, R. (2018). Emicizumab Prophylaxis in Patients Who Have Hemophilia A without Inhibitors. The New England journal of medicine, 379(9), 811–822. https://doi.org/10.1056/NEJMoa1803550
- Chaudhry R, Killeen RB, Babiker HM. Physiology, Coagulation Pathways. [Updated 2025 Jun 2]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2026 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482253/
- Starikova, E. A., Mammedova, J. T., Rubinstein, A. A., Sokolov, A. V., & Kudryavtsev, I. V. (2025). Activation of the Coagulation Cascade as a Universal Danger Sign. Current issues in molecular biology, 47(2), 108. https://doi.org/10.3390/cimb47020108
- Lenting, P. J., Christophe, O. D., & Denis, C. V. (2015). von Willebrand factor biosynthesis, secretion, and clearance: connecting the far ends. Blood, 125(13), 2019–2028. https://doi.org/10.1182/blood-2014-06-528406
- Puy, C., Rigg, R. A., & McCarty, O. J. (2016). The hemostatic role of factor XI. Thrombosis research, 141 Suppl 2(Suppl 2), S8–S11. https://doi.org/10.1016/S0049-3848(16)30354-1
- Srivastava, A., Rangarajan, S., Kavakli, K., Klamroth, R., Kenet, G., Khoo, L., You, C. W., Xu, W., Malan, N., Frenzel, L., Bagot, C. N., Stasyshyn, O., Chang, C. Y., Poloskey, S., Qiu, Z., Andersson, S., Mei, B., & Pipe, S. W. (2023). Fitusiran prophylaxis in people with severe haemophilia A or haemophilia B without inhibitors (ATLAS-A/B): a multicentre, open-label, randomised, phase 3 trial. The Lancet. Haematology, 10(5), e322–e332. https://doi.org/10.1016/S2352-3026(23)00037-6
- Gailani, D., & Gruber, A. (2024). Targeting factor XI and factor XIa to prevent thrombosis. Blood, 143(15), 1465–1475. https://doi.org/10.1182/blood.2023020722
- Eikelboom, J., Floege, J., Thadhani, R., Weitz, J. I., & Winkelmayer, W. C. (2021). Anticoagulation in patients with kidney failure on dialysis: factor XI as a therapeutic target. Kidney international, 100(6), 1199–1207. https://doi.org/10.1016/j.kint.2021.08.028