Key Takeaways
Myelodysplastic syndrome (MDS) is a group of bone marrow cancers in which mutated stem cells produce defective blood cells. Most are destroyed before reaching the bloodstream, causing low blood counts.
- Signs and symptoms ▾: Patients usually present with the consequences of bone marrow failure: fatigue and pallor from anemia, infections from neutropenia, and easy bruising or bleeding from thrombocytopenia.
- Key Laboratory Investigations ▾: Diagnosis requires a complete blood count, peripheral blood smear, bone marrow biopsy showing dysplasia in at least 10% of one cell lineage with under 20% blasts, plus cytogenetics and next-generation sequencing (NGS) [3,5].
- Treatment & Management ▾: Treatment depends on risk. Lower-risk patients receive ESAs, luspatercept, lenalidomide (for del(5q)), or imetelstat (approved 2024) to manage anemia [6,7,10]. Higher-risk patients receive hypomethylating agents and, when possible, an allogeneic stem cell transplant — the only curative therapy [8].
*Click ▾ for more information
Introduction
Imagine a busy factory that keeps assembling products, but most of them fail quality control before they ever leave the building. That is what happens inside the bone marrow in myelodysplastic syndrome. The marrow is full, but the patient's blood counts are low. This contradiction sits at the heart of the disease.
Myelodysplastic syndrome (MDS) is a group of clonal stem cell cancers in which acquired mutations stop blood-forming cells from maturing properly. The cells that are produced look abnormal under the microscope (this is dysplasia) and most die in the marrow before reaching the circulation. The result is ineffective hematopoiesis — production without delivery [3].
MDS is mostly a disease of older adults, with a peak incidence after age 70 [3]. About a third of patients eventually progress to acute myeloid leukemia (AML), so accurate diagnosis and risk-based treatment are central to care.
This article walks through the causes, signs and symptoms, classification, laboratory workup, prognosis, and treatment of myelodysplastic syndrome.
Myelodysplastic Syndrome (MDS) Symptoms
Symptoms develop slowly and are easy to dismiss at first. Most patients present with the effects of one or more cytopenias.
- Anemia (low red cells): fatigue, breathlessness on exertion, pallor, dizziness. Anemia is the most common finding.
- Neutropenia (low neutrophils): recurrent or unusual infections, especially of the skin, mouth, and chest.
- Thrombocytopenia (low platelets): easy bruising, petechiae (pinpoint red spots), nosebleeds, or bleeding gums.
Some patients also have nonspecific symptoms such as weight loss or low-grade fever. In an older adult with persistent macrocytic anemia and no obvious cause, MDS should be on the differential.
Causes and Risk Factors
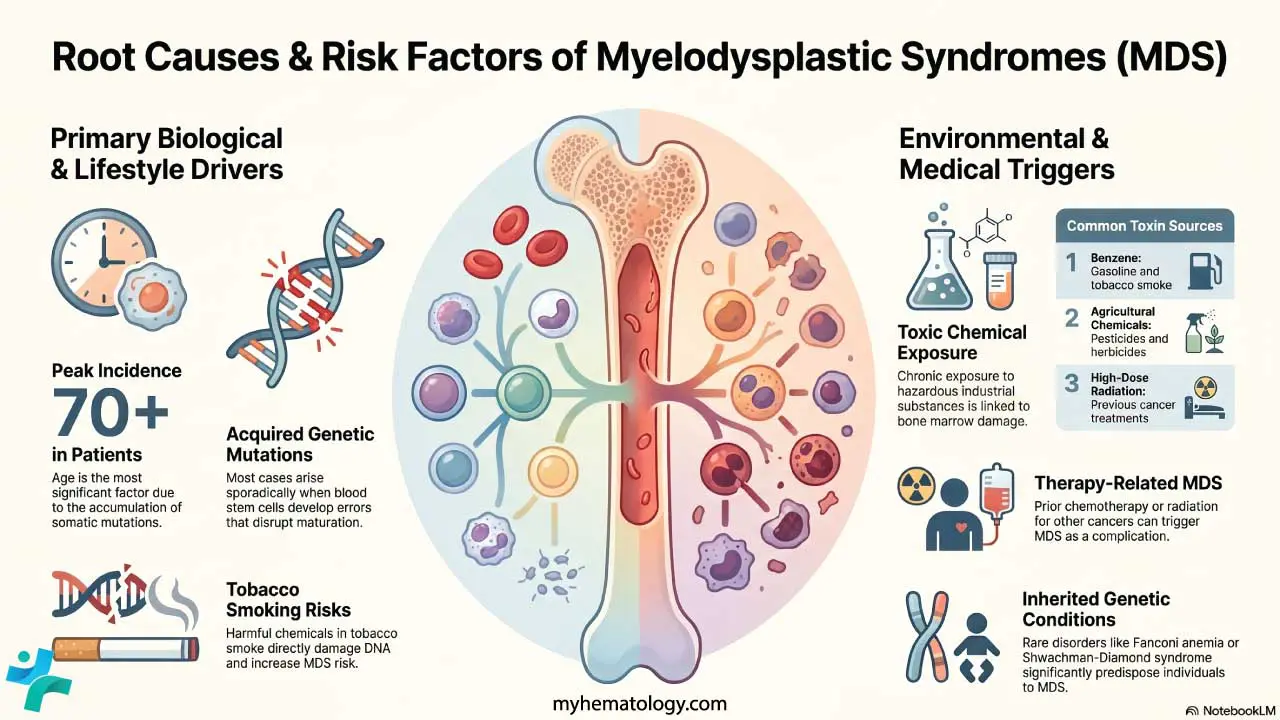
MDS begins when a single blood-forming stem cell acquires a damaging mutation and gives rise to a clone of dysfunctional descendants. Anything that increases DNA damage in marrow stem cells increases MDS risk.
The leading risk factor is age. Mutations build up in stem cells over a lifetime, and by the seventh decade the chance of a clinically significant clone rises sharply [3]. On top of this background, several specific exposures matter:
- Previous chemotherapy or radiation. Alkylating agents and topoisomerase II inhibitors are well-known culprits in therapy-related MDS, which usually appears 2–10 years after treatment.
- Benzene. Found in industrial solvents, gasoline, and tobacco smoke. A long-established marrow toxin.
- Tobacco smoking. Independently raises MDS risk through repeated DNA damage.
- Pesticides and herbicides. Less well established than benzene, but consistently linked in occupational studies.
- Inherited bone marrow failure syndromes. Rare causes in younger patients, including Fanconi anemia, Diamond-Blackfan anemia, and Shwachman-Diamond syndrome.
In many older patients, no specific exposure is found. The mutations have simply accumulated with age.
A precursor state called clonal hematopoiesis of indeterminate potential (CHIP) is now recognized: a person carries an MDS-type mutation in their stem cells but has normal blood counts. Most people with CHIP never develop MDS, but the risk is higher than baseline, and CHIP is now part of the diagnostic conversation [3].
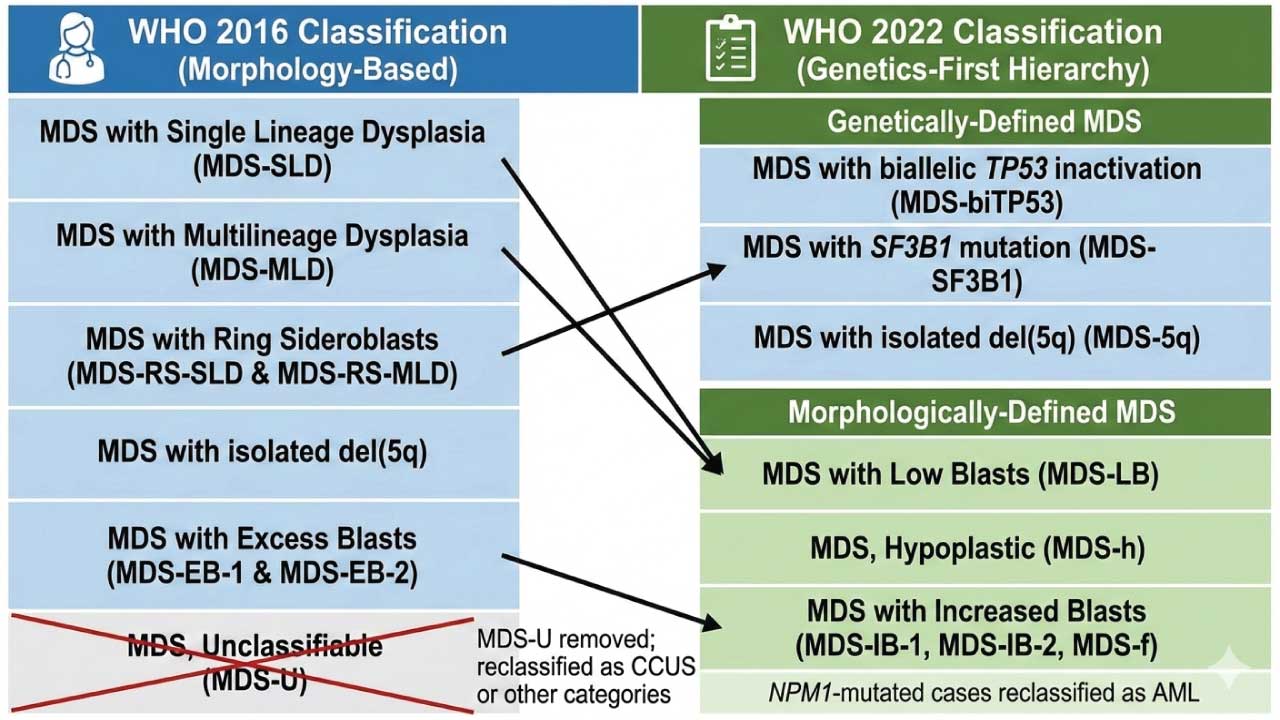
WHO and ICC 2022 Classifications
In 2022, two major classifications of myeloid neoplasms were published in parallel: the 5th edition WHO Classification (WHO-HAEM5) [3] and the International Consensus Classification (ICC) [4]. Both renamed MDS as "myelodysplastic neoplasms" to underline that this is a true neoplastic process. They also moved the field decisively toward genetic definitions.
The WHO-HAEM5 splits MDS into two branches.
Genetically defined MDS
If one of three defining genetic abnormalities is present, the patient is classified by genetics, regardless of dysplastic appearance:
- MDS with biallelic TP53 inactivation (MDS-biTP53) — two or more TP53 mutations, or one mutation with loss of the other allele (for example, a 17p deletion). This subtype carries the worst prognosis.
- MDS with SF3B1 mutation (MDS-SF3B1) — replaces the older "MDS with ring sideroblasts" category. The mutation now defines the entity; the iron rings are a surrogate when SF3B1 is not tested.
- MDS with isolated del(5q) (MDS-5q) — broadly similar to before, but a TP53 mutation must now be excluded because it changes management.
Morphologically defined MDS
If no defining genetic abnormality is found, classification depends on blast count and marrow cellularity:
- MDS with low blasts (MDS-LB): under 5% marrow blasts and under 2% blood blasts.
- MDS, hypoplastic (MDS-h): age-adjusted marrow cellularity under 25%. Often immune-mediated and may respond to immunosuppression.
- MDS with increased blasts (MDS-IB): replaces "excess blasts." Subdivided into MDS-IB1 (5–9% marrow or 2–4% blood blasts) and MDS-IB2 (10–19% marrow or 5–19% blood blasts, or any Auer rods).
- MDS with fibrosis (MDS-f): increased blasts plus grade 2 or 3 marrow fibrosis. Aggressive course, worse survival.
The ICC system uses similar logic but creates an "MDS/AML" overlap category at 10–19% blasts, rather than placing all of these cases inside MDS [4].
Laboratory Investigations
The diagnostic workup of myelodysplastic syndrome moves from initial screening, to bone marrow examination, to molecular profiling, with exclusionary tests run alongside.
Initial screening: CBC and peripheral blood smear
Every workup starts with a complete blood count (CBC) and peripheral blood film (PBF). The CBC defines the cytopenias. The smear gives the first dysplastic clues.
- Persistent cytopenias lasting at least 4–6 months. Modern WHO/ICC criteria use Hb under 10 g/dL, ANC under 1.8 × 10⁹/L, and platelets under 150 × 10⁹/L as practical thresholds [3,4].
- Macrocytosis. Mean cell volume over 100 fL with oval macrocytes is a classic early sign.
- Dysgranulopoiesis. Look for Pseudo-Pelger-Huët anomaly (neutrophils with bilobed, "pince-nez" nuclei) and hypogranular cytoplasm.
- Circulating blasts. Even 1% blasts in the blood is a red flag for higher-risk MDS.
The gold standard: bone marrow biopsy
A bone marrow aspirate plus trephine biopsy confirms the diagnosis and excludes mimics.
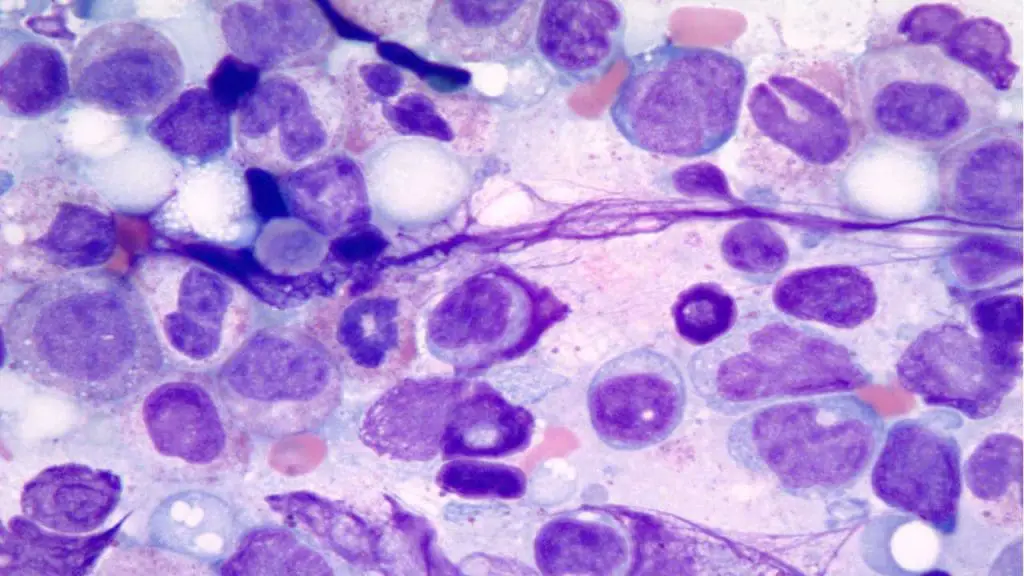
- Dysplasia must be present in at least 10% of cells in at least one lineage:
- Erythroid line: nuclear budding, internuclear bridging, ring sideroblasts.
- Myeloid line: hyposegmentation, hypogranulation.
- Megakaryocytic line: micromegakaryocytes (small cells with single or "pawn-broker" nuclei) — the most specific marker.
- Prussian blue (iron) stain identifies ring sideroblasts. A ring sideroblast has iron-loaded mitochondria encircling at least one-third of the nucleus.
- Blast count distinguishes MDS (under 20%) from AML (20% or more).
- CD34 immunohistochemistry highlights ALIP (abnormal localization of immature precursors), which suggests higher risk of AML transformation.
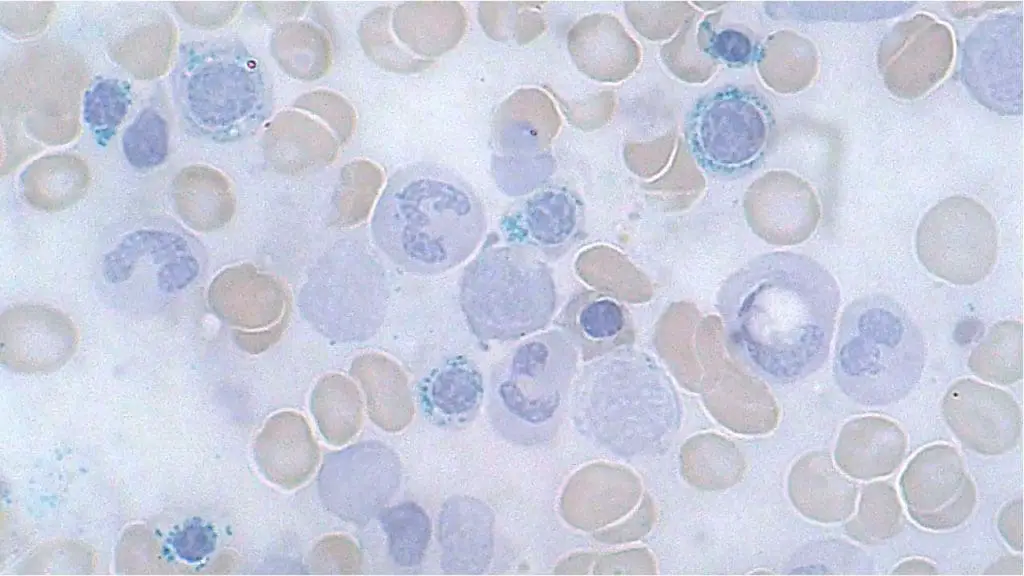
Molecular and cytogenetic profiling
A modern MDS diagnosis is incomplete without genetic data.
- Conventional karyotyping picks up large chromosomal changes such as del(5q), trisomy 8, or monosomy 7. About half of patients have a normal karyotype, which is why molecular testing is essential.
- Next-generation sequencing (NGS) of a myeloid gene panel identifies driver mutations. SF3B1, ASXL1, and TET2 support a clonal diagnosis. TP53 (especially biallelic) and RUNX1 signal an aggressive course.
- The IPSS-M integrates these mutations with clinical data for the most accurate risk stratification available [2].
Furthermore, the use of peripheral blood next-generation sequencing (often referred to as liquid biopsies) is increasingly utilized in modern practice. While a bone marrow biopsy remains the gold standard for the initial morphological diagnosis, analyzing cell-free DNA (cfDNA) in the blood is becoming a valuable, less invasive tool to track clonal dynamics, assess minimal residual disease (MRD), and monitor disease progression over time without the constant need for repeated marrow aspirations [12].
Ancillary and exclusionary tests
MDS is partly a diagnosis of exclusion. Several reversible conditions can mimic dysplasia:
- Vitamin B12 and folate deficiency can perfectly imitate MDS morphology.
- Copper deficiency (often from excessive zinc supplementation) causes vacuolated precursors and ring sideroblasts.
- Reticulocyte count is typically low in MDS, reflecting ineffective hematopoiesis.
- Flow cytometry can support borderline cases by detecting aberrant marker expression on myeloid cells (loss of CD13/CD11b, gain of CD7).
VEXAS syndrome
VEXAS syndrome (caused by somatic UBA1 mutations) must be excluded, particularly in older male patients presenting with macrocytic anemia, vacuoles in marrow precursor cells, and unexplained systemic inflammation (such as relapsing polychondritis, skin lesions, or recurrent fevers). Its bone marrow morphology can closely mimic MDS, but standard genetic testing via peripheral blood or marrow for the UBA1 gene is definitive [11].
Prognosis: The IPSS-M
The Molecular International Prognostic Scoring System (IPSS-M) was published by Bernard and colleagues in 2022 and is now considered the most accurate risk tool for MDS [2]. It builds on the older IPSS-R by adding genetic data. Where IPSS-R used only blood counts and chromosomes, IPSS-M layers in mutations in 31 genes.
This is not a small change. In the original IPSS-M cohort, around 46% of patients were reassigned to a different risk category compared with IPSS-R. About 24% moved up (often because of a hidden TP53 or ASXL1 mutation) and about 22% moved down (often because of an isolated SF3B1 mutation) [2].
The three pillars of IPSS-M
- Clinical parameters: hemoglobin, platelet count, marrow blast percentage.
- Cytogenetic risk: the five-tier IPSS-R cytogenetic categories (Very Good to Very Poor).
- Molecular data: presence and variant allele frequency (VAF) of mutations in 31 specific genes. TP53 status is split into "single-hit" and "multi-hit" (biallelic), because the latter is much more aggressive.
The 31 Genes of the IPSS-M
These genes are selected for their independent impact on prognosis. For clinical purposes, they are often grouped by their biological function:
The six IPSS-M risk strata
- Very Low
- Low
- Moderate Low
- Moderate High
- High
- Very High
A practical caveat
NGS panels are still not universally available, and many centers around the world continue to use the IPSS-R. The IPSS-M is the reference standard, but the IPSS-R is far from obsolete in routine practice.
Myelodysplastic Syndrome (MDS) Treatment
Treatment is built around the IPSS-M risk score, age, comorbidities, and patient goals. The strategic split is clear: in lower-risk MDS, the priority is quality of life and managing anemia. In higher-risk MDS, the priority is delaying progression to AML and, where possible, cure.
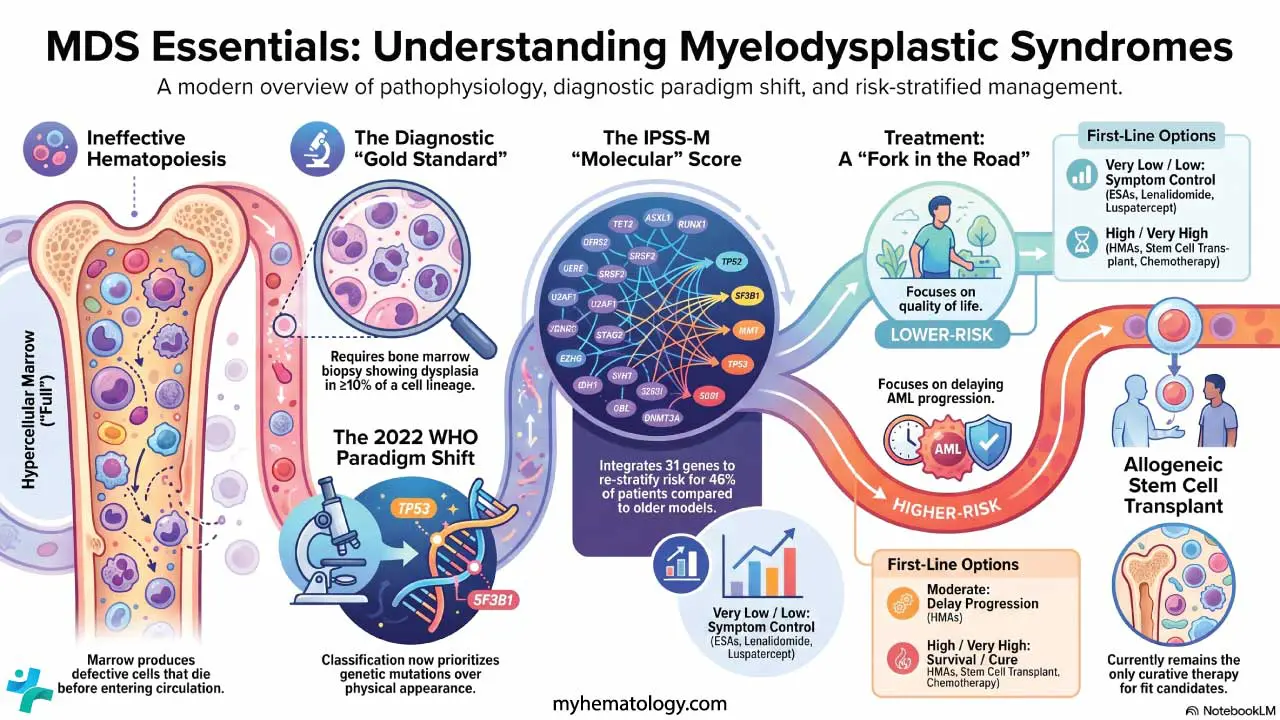
Lower-risk MDS
The dominant clinical problem is chronic transfusion-dependent anemia.
- First-line (Low EPO): Erythropoiesis-stimulating agents (ESAs) such as epoetin alfa or darbepoetin remain the frontline choice for symptomatic anemia, provided endogenous erythropoietin is below 500 U/L [10].
- First-line (Alternative): Luspatercept, an erythroid maturation agent, is now favored as first-line therapy (ahead of ESAs) for lower-risk MDS patients who are transfusion-dependent, particularly those with ring sideroblasts or SF3B1mutations. The COMMANDS trial demonstrated its superiority over epoetin alfa in achieving transfusion independence in this cohort [6].
- Second-line: Imetelstat (Rytelo), a first-in-class telomerase inhibitor approved by the FDA in June 2024, is indicated for adults with low- to intermediate-1 risk MDS with transfusion-dependent anemia (≥4 RBC units over 8 weeks) who have failed, lost response to, or are ineligible for ESAs [7,10].
- Targeted Deletion: Lenalidomide remains the standard of care specifically for MDS with isolated del(5q) [10].
- Hypoplastic MDS: Immunosuppressive therapy (IST), such as antithymocyte globulin plus cyclosporine, can help younger patients where T-cell-mediated destruction contributes to marrow failure.
Higher-risk MDS
The marrow is failing, and the risk of AML is rising.
- Hypomethylating agents (HMAs) — azacitidine and decitabine — are the backbone of therapy. Azacitidine has shown an overall survival benefit in randomized trials [8].
- HMA + venetoclax. Borrowed from acute myeloid leukemia (AML), this combination is used in fit, higher-risk patients to deepen responses. However, unlike in AML, prolonged venetoclax utilization in MDS causes profound and prolonged myelosuppression. Management strictly requires dose and duration adjustments, for example, often reducing venetoclax to 14 days or fewer per cycle alongside the aggressive use of growth factors to prevent severe infectious complications [13].
- Intensive induction chemotherapy (7+3 style) is reserved for fit, younger patients with high blast counts, often as a bridge to transplant.
Allogeneic stem cell transplantation: the only curative option
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) remains the only therapy that can cure MDS. For lower-risk patients, transplant is usually deferred because the procedure carries significant morbidity and mortality. For higher-risk patients (Intermediate to Very High on IPSS-M), transplant is pursued as soon as a suitable donor is found and the patient is fit enough to proceed [8].
Supportive care for everyone
Supportive care underpins management at every risk level.
- Transfusion support with red cells and platelets, used judiciously.
- Iron chelation with deferasirox or deferoxamine to prevent iron overload damage to the heart and liver in chronically transfused patients with a long life expectancy.
- Antibiotic prophylaxis in patients with severe, persistent neutropenia.
Targeted therapies
Mutation-directed therapy is expanding, with IDH1 and IDH2 inhibitors (ivosidenib, enasidenib) used off-label or in trials for patients with these mutations. Conversely, managing TP53-mutated MDS (particularly the multi-hit/biallelic subtype) remains a major clinical challenge, especially following the discontinuation of the CD47-targeted antibody magrolimab in early 2024 due to futility and increased mortality. Because standard HMAs offer very short-lived responses, immediate evaluation for allogeneic stem cell transplantation (if fit) or enrollment in clinical trials investigating novel immunotherapies is the recommended standard of care for multi-hit TP53 MDS [14].
Treatment by Risk Category
| Risk group | Primary goal | First-line options |
|---|---|---|
| Very low / low | Symptom control | ESAs, luspatercept, lenalidomide (del(5q)), imetelstat (post-ESA), IST (hypoplastic) |
| Moderate | Delay progression | HMAs, clinical trials |
| High / very high | Survival or cure | HMAs, allogeneic HSCT, intensive chemotherapy |
Frequently Asked Questions (FAQs)
What is myelodysplastic syndrome (MDS) in simple terms?
MDS is a group of bone marrow cancers in which mutated stem cells produce defective blood cells. The marrow is busy but unproductive: it makes plenty of cells, but most die before reaching the bloodstream. Patients develop low red cells, white cells, or platelets, and around one in three eventually progress to AML.
Who gets MDS? MDS is mainly a disease of older adults, with peak incidence after age 70. It also appears in younger people who have had chemotherapy or radiation for an earlier cancer (therapy-related MDS), in those with long-term exposure to benzene or tobacco smoke, and rarely in people with inherited bone-marrow failure syndromes.
How is MDS diagnosed? A complete blood count showing persistent cytopenias, a peripheral blood smear, and a bone marrow biopsy are required. The biopsy must show dysplasia in at least 10% of cells in one lineage and fewer than 20% blasts. Cytogenetics (karyotype) and next-generation sequencing of a myeloid panel are now standard, because mutations directly affect risk score and treatment.
Is MDS curable? Drug therapy alone does not cure MDS. The only curative treatment is an allogeneic stem cell transplant. Because transplants carry significant risks, they are usually reserved for fitter patients with higher-risk disease. Lower-risk patients are managed to maintain quality of life and delay progression.
What is the difference between MDS and AML? The key difference is the percentage of blasts in the bone marrow. MDS is defined by under 20% blasts; AML is 20% or more. About a third of MDS patients eventually progress to AML, particularly those in higher-risk IPSS-M categories.
Why is the bone marrow "full" if blood counts are low? Because of ineffective hematopoiesis. The marrow produces many cell precursors, but most undergo apoptosis (programmed cell death) before they leave the marrow. The factory is running, but most products fail quality control.
What is new in MDS treatment? Three updates stand out. Imetelstat (Rytelo) was approved in June 2024 for transfusion-dependent anemia in lower-risk MDS that has failed ESAs. Luspatercept was expanded in 2023 to first-line lower-risk anemia regardless of ring-sideroblast status. Magrolimab development for hematologic cancers was discontinued in 2024 after trials showed futility and increased mortality.
Glossary of Related Medical Terms
- Allogeneic stem cell transplant (allo-HSCT): A transplant in which healthy stem cells from a matched donor replace the patient's diseased bone marrow. The only curative MDS therapy.
- Apoptosis: Programmed cell death. Excess apoptosis of marrow precursors explains why patients with MDS have a "full" marrow but low blood counts.
- Auer rod: Pink, needle-shaped inclusion in a myeloid blast. If seen in MDS, the disease is automatically high-risk.
- Blast: An immature precursor cell. Healthy marrow has under 5% blasts; MDS has under 20%; ≥20% defines AML.
- Clonal: Originating from a single mutated parent cell.
- Clonal hematopoiesis of indeterminate potential (CHIP): A pre-MDS state with a cancer-related mutation but normal blood counts.
- Cytopenia: A low blood-cell count: anemia (red cells), neutropenia (neutrophils), or thrombocytopenia (platelets).
- Dysplasia: Abnormal cell shape, size, or organization under the microscope. The morphological hallmark of MDS.
- Erythropoiesis-stimulating agent (ESA): A drug that mimics natural erythropoietin and stimulates red-cell production.
- Hematopoiesis: The production of new blood cells in the bone marrow.
- Hypomethylating agent (HMA): Drugs such as azacitidine and decitabine that reactivate silenced anti-cancer genes.
- Imetelstat: A telomerase inhibitor approved in 2024 for transfusion-dependent anemia in lower-risk MDS after ESA failure.
- IPSS-M: Molecular International Prognostic Scoring System. Combines blood counts, chromosomes, and 31-gene NGS to predict outcomes.
- Karyotype: A picture of all of a cell's chromosomes used to detect large changes such as del(5q) or monosomy 7.
- Lenalidomide: Standard treatment for MDS with isolated del(5q).
- Luspatercept: An erythroid maturation agent that helps red-cell precursors mature and reduces transfusion needs in lower-risk MDS.
- Macrocytosis: Larger-than-normal red cells (MCV > 100 fL); a common early sign of MDS.
- Next-generation sequencing (NGS): DNA-sequencing technology that reads many genes at once.
- Pancytopenia: Low counts in all three blood lineages simultaneously.
- Pseudo-Pelger-Huët anomaly: Neutrophils with bilobed, "pince-nez" nuclei. A classic dysplastic feature.
- Ring sideroblast: A red-cell precursor with a ring of iron-loaded mitochondria around its nucleus. Strongly linked to SF3B1-mutated MDS.
- Somatic mutation: A mutation acquired during life rather than inherited.
- TP53: A tumor-suppressor gene. Biallelic TP53 damage marks the most aggressive MDS subtype.
Disclaimer: This article is intended for educational and informational purposes only. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Greenberg, P. L., Tuechler, H., Schanz, J., Sanz, G., Garcia-Manero, G., Solé, F., Bennett, J. M., Bowen, D., Fenaux, P., Dreyfus, F., Kantarjian, H., Kuendgen, A., Levis, A., Malcovati, L., Cazzola, M., Cermak, J., Fonatsch, C., Le Beau, M. M., Slovak, M. L., Krieger, O., … Haase, D. (2012). Revised international prognostic scoring system for myelodysplastic syndromes. Blood, 120(12), 2454–2465. https://doi.org/10.1182/blood-2012-03-420489
- Bernard, E., Tuechler, H., Greenberg, P. L., Hasserjian, R. P., Arango Ossa, J. E., Nannya, Y., Devlin, S. M., Creignou, M., Pinel, P., Monnier, L., Gundem, G., Medina-Martinez, J. S., Domenico, D., Jädersten, M., Germing, U., Sanz, G., van de Loosdrecht, A. A., Kosmider, O., Follo, M. Y., Thol, F., … Papaemmanuil, E. (2022). Molecular International Prognostic Scoring System for Myelodysplastic Syndromes. NEJM evidence, 1(7), EVIDoa2200008. https://doi.org/10.1056/EVIDoa2200008
- Khoury, J. D., Solary, E., Abla, O., Akkari, Y., Alaggio, R., Apperley, J. F., Bejar, R., Berti, E., Busque, L., Chan, J. K. C., Chen, W., Chen, X., Chng, W. J., Choi, J. K., Colmenero, I., Coupland, S. E., Cross, N. C. P., De Jong, D., Elghetany, M. T., Takahashi, E., … Hochhaus, A. (2022). The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia, 36(7), 1703–1719. https://doi.org/10.1038/s41375-022-01613-1
- Arber, D. A., Orazi, A., Hasserjian, R. P., Borowitz, M. J., Calvo, K. R., Kvasnicka, H. M., Wang, S. A., Bagg, A., Barbui, T., Branford, S., Bueso-Ramos, C. E., Cortes, J. E., Dal Cin, P., DiNardo, C. D., Dombret, H., Duncavage, E. J., Ebert, B. L., Estey, E. H., Facchetti, F., Foucar, K., … Tefferi, A. (2022). International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood, 140(11), 1200–1228. https://doi.org/10.1182/blood.2022015850
- Garcia-Manero G. (2023). Myelodysplastic syndromes: 2023 update on diagnosis, risk-stratification, and management. American journal of hematology, 98(8), 1307–1325. https://doi.org/10.1002/ajh.26984
- Platzbecker, U., Della Porta, M. G., Santini, V., Zeidan, A. M., Komrokji, R. S., Shortt, J., Valcarcel, D., Jonasova, A., Dimicoli-Salazar, S., Tiong, I. S., Lin, C. C., Li, J., Zhang, J., Giuseppi, A. C., Kreitz, S., Pozharskaya, V., Keeperman, K. L., Rose, S., Shetty, J. K., Hayati, S., … Garcia-Manero, G. (2023). Efficacy and safety of luspatercept versus epoetin alfa in erythropoiesis-stimulating agent-naive, transfusion-dependent, lower-risk myelodysplastic syndromes (COMMANDS): interim analysis of a phase 3, open-label, randomised controlled trial. Lancet (London, England), 402(10399), 373–385. https://doi.org/10.1016/S0140-6736(23)00874-7
- Platzbecker, U., Santini, V., Fenaux, P., Sekeres, M. A., Savona, M. R., Madanat, Y. F., Díez-Campelo, M., Valcárcel, D., Illmer, T., Jonášová, A., Bělohlávková, P., Sherman, L. J., Berry, T., Dougherty, S., Shah, S., Xia, Q., Sun, L., Wan, Y., Huang, F., Ikin, A., … Zeidan, A. M. (2024). Imetelstat in patients with lower-risk myelodysplastic syndromes who have relapsed or are refractory to erythropoiesis-stimulating agents (IMerge): a multinational, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet (London, England), 403(10423), 249–260. https://doi.org/10.1016/S0140-6736(23)01724-5
- Kröger N. (2025). Treatment of high-risk myelodysplastic syndromes. Haematologica, 110(2), 339–349. https://doi.org/10.3324/haematol.2023.284946
- Merz, A. M. A., & Platzbecker, U. (2025). Treatment of lower-risk myelodysplastic syndromes. Haematologica, 110(2), 330–338. https://doi.org/10.3324/haematol.2023.284945
- McMahon, C., Raddi, M. G., Mohan, S., & Santini, V. (2025). New Approvals in Low- and Intermediate-Risk Myelodysplastic Syndromes. American Society of Clinical Oncology educational book. American Society of Clinical Oncology. Annual Meeting, 45(3), e473654. https://doi.org/10.1200/EDBK-25-473654
- Beck, D. B., Ferrada, M. A., Sikora, K. A., Ombrello, A. K., Collins, J. C., Pei, W., Balanda, N., Ross, D. L., Ospina Cardona, D., Wu, Z., Patel, B., Manthiram, K., Groarke, E. M., Gutierrez-Rodrigues, F., Hoffmann, P., Rosenzweig, S., Nakabo, S., Dillon, L. W., Hourigan, C. S., Tsai, W. L., … Grayson, P. C. (2020). Somatic Mutations in UBA1 and Severe Adult-Onset Autoinflammatory Disease. The New England journal of medicine, 383(27), 2628–2638. https://doi.org/10.1056/NEJMoa2026834
- Colmenares, R., Álvarez, N., Barrio, S., Martínez-López, J., & Ayala, R. (2022). The Minimal Residual Disease Using Liquid Biopsies in Hematological Malignancies. Cancers, 14(5), 1310. https://doi.org/10.3390/cancers14051310
- El-Cheikh, J., Bidaoui, G., Saleh, M., Moukalled, N., Abou Dalle, I., & Bazarbachi, A. (2023). Venetoclax: A New Partner in the Novel Treatment Era for Acute Myeloid Leukemia and Myelodysplastic Syndrome. Clinical hematology international, 5(2-3), 143–154. https://doi.org/10.1007/s44228-023-00041-x
- Albakri M. M. (2025). TP53-mutated MDS and AML: immune dysregulation, tumor microenvironment, and emerging therapeutic strategies. Frontiers in oncology, 15, 1655486. https://doi.org/10.3389/fonc.2025.1655486