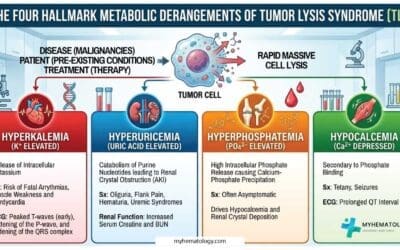
TL;DR
Essential thrombocythemia (ET) is a chronic blood cancer characterized by an abnormal increase in platelet production by the bone marrow, leading to an increased risk of blood clots (thrombosis) and bleeding complications.
- Pathogenesis ▾:Primarily driven by mutations in JAK2, CALR, or MPL genes, disrupting the JAK-STAT pathway and promoting uncontrolled megakaryocyte (platelet precursor) growth.
- Symptoms ▾: Often asymptomatic, detected incidentally during routine blood tests. Potential symptoms include:
- Thrombotic events (e.g., deep vein thrombosis, stroke)
- Hemorrhagic events (less common)
- Constitutional symptoms (e.g., fatigue, headache)
- Splenomegaly (enlarged spleen)
- Complications ▾:
- Myelofibrosis transformation
- Acute myeloid leukemia (AML) (rare)
- Venous thromboembolism (VTE)
- Arterial thrombosis
- Bleeding risks
- Diagnosis ▾:
- Elevated platelet count (> 450,000/µL)
- Bone marrow examination (no myelofibrosis)
- Absence of other clonal myeloid disease
- Presence of JAK2, CALR, or MPL mutation (at least one)
- Treatment ▾: Low-risk vs. high-risk stratification guides treatment decisions. Options include:
- Cytoreductive therapy (e.g., hydroxyurea) – high-risk patients
- Antiplatelet therapy (e.g., aspirin) – low-risk patients and those with thrombosis history
- Other modalities: interferon alfa, JAK inhibitors, splenectomy (rarely)
- Prognosis ▾: With proper management, individuals with ET can have a near-normal lifespan. Regular follow-up is essential for monitoring treatment response, detecting complications early, and optimizing long-term health.
- Differential diagnosis ▾: Reactive thrombocytosis is the most common cause, often due to underlying conditions like infections or inflammation. Other possibilities include other myeloproliferative neoplasms (MPNs) like chronic myeloid leukemia (CML) and primary myelofibrosis (PMF).
*Click ▾ for more information
What is Essential Thrombocythemia (ET)?
Essential thrombocythemia (ET) is a chronic blood cancer characterized by an abnormal increase in the production of platelets by the bone marrow. These platelets are involved in blood clotting, and their excessive number can lead to increased risk of blood clots and bleeding complications.
Essential thrombocythemia (ET) belongs to a group of blood disorders known as myeloproliferative neoplasms (MPNs). These disorders are characterized by the clonal (abnormal) growth and proliferation of blood cell precursors in the bone marrow. In ET, the specific cell line affected is megakaryocytes, which are responsible for producing platelets.
While classified as a cancer, essential thrombocythemia (ET) typically has a slow progression and good prognosis with proper management.
Pathogenesis and Pathophysiology

Myeloproliferation and the JAK-STAT pathway
Essential thrombocythemia (ET) is driven by abnormal cell signaling within the bone marrow, leading to uncontrolled proliferation of megakaryocytes and excessive platelet production. This malfunction primarily involves the JAK-STAT pathway:
- JAK (Janus Kinase): An enzyme that initiates signaling within the cell after receiving signals from outside.
- STAT (Signal Transducer and Activator of Transcription): A protein that, once activated by JAK, travels to the nucleus and influences gene expression.
Role of JAK2, CALR and MPL mutations in abnormal cell signaling

Mutations in JAK2, CALR, MPL, and JAK2 exon 12: These mutations are the hallmark of essential thrombocythemia (ET) and are found in around 90% of patients. They affect different parts of the JAK-STAT pathway, leading to abnormal cell signaling.
- JAK2 (Janus Kinase 2) V617F mutation: This is the most common driver mutation, found in approximately 50-60% of ET patients. It leads to constitutive activation of the JAK-STAT signaling pathway, making the hematopoietic cells hypersensitive to growth factors like thrombopoietin (TPO). The V617F mutation is a point mutation in the pseudokinase domain of JAK2, disrupting its inhibitory function and leading to uncontrolled signaling.
- CALR (Calreticulin) mutations: These are the second most frequent driver mutations, found in about 20-30% of ET patients who are JAK2 V617F-negative. Most CALR mutations are insertions or deletions in exon 9, resulting in a novel C-terminus of the calreticulin protein. This mutant calreticulin interacts with the MPL receptor, leading to its activation and downstream JAK-STAT signaling, even in the absence of TPO. Different types of CALR mutations (Type 1 and Type 2 being the most common) may be associated with slightly different clinical phenotypes.
- MPL (Myeloproliferative Leukemia Virus Oncogene) mutations: These mutations, primarily in exon 10 (e.g., W515L, W515K), occur in a smaller percentage (2-5%) of ET patients who are negative for both JAK2 and CALR mutations. MPL encodes the thrombopoietin receptor (TpoR). These mutations typically lead to ligand-independent dimerization and activation of the MPL receptor, again resulting in constitutive activation of the JAK-STAT pathway.
- “Triple-Negative” ET: In the 10-20% of ET patients who lack detectable mutations in JAK2, CALR, and MPL, the underlying genetic drivers are still largely unknown. Research is ongoing to identify other potential mutations or mechanisms responsible for the disease in these individuals. Some studies have identified other less frequent mutations in genes like THPO or EZH2 in a small subset of these patients, but a clear driver is yet to be consistently identified. This suggests that there may be other as-yet-undiscovered genetic alterations or even non-genetic mechanisms involved in the pathogenesis of ET in these cases.
Disruption of normal hematopoiesis leading to increased platelet production
These mutations in JAK2, CALR, or MPL lead to dysregulation of the JAK-STAT pathway, which normally controls cell growth, differentiation, and survival. This dysregulation disrupts the delicate balance of blood cell production in the bone marrow, favoring uncontrolled megakaryocyte proliferation and increased platelet release.
Epidemiology and risk factors
Incidence and Prevalence
- Incidence: Estimated to be 0.2 to 2.5 cases per 100,000 people annually, meaning it is a relatively rare disease.
- Prevalence: Estimated to be 38 to 57 cases per 100,000 people, suggesting more individuals are diagnosed with the condition than are newly diagnosed each year. However, due to the often mild symptoms, some cases may remain undiagnosed, potentially impacting the actual prevalence.
Age, Gender, and Ethnicity
- Age: Essential thrombocythemia (ET) typically occurs in adults aged 50-60 years, but it can affect any age group, including children.
- Gender: Women are slightly more likely to be diagnosed with essential thrombocythemia (ET) than men, with a female-to-male ratio of approximately 2:1.
- Ethnicity: While essential thrombocythemia (ET) can occur in any ethnic group, certain populations, such as East Asians, may have a lower prevalence compared to others.
Familial Predisposition
- Family history of essential thrombocythemia (ET) is a minor risk factor, suggesting a potential genetic component to the disease in some cases. However, most cases occur sporadically without a clear family link.
Essential Thrombocythemia (ET) Symptoms
Many individuals with ET, particularly in the early stages, may be asymptomatic. The condition is often discovered incidentally during a routine blood test done for another reason. However, as the disease progresses or with very high platelet counts, various signs and symptoms can develop. These are primarily related to two main issues caused by the increased and often dysfunctional platelets: blood clots (thrombosis) and, less commonly, bleeding (hemorrhage).
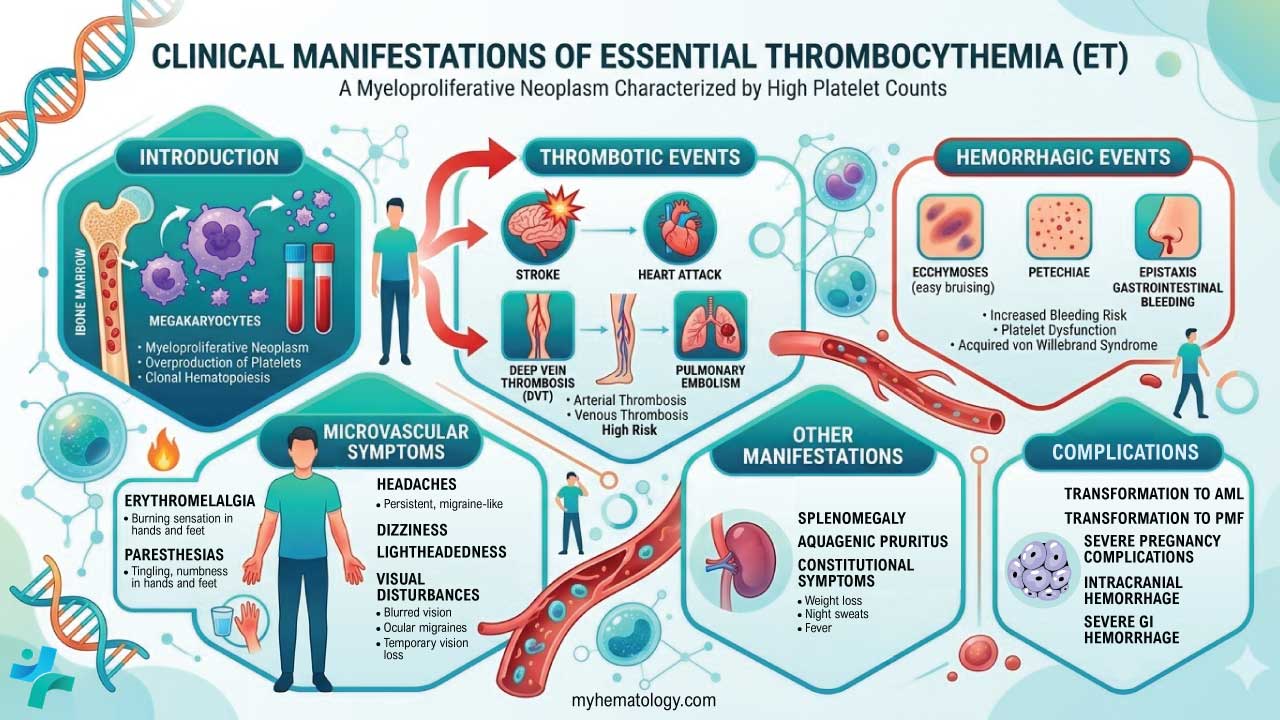
Symptoms Related to Blood Clots (Thrombosis)
The increased number of platelets can make the blood “stickier” and more prone to forming clots in blood vessels throughout the body. The symptoms depend on the location of the clot.
Small Blood Vessel Issues (Microvascular Disturbances)
- Erythromelalgia: Burning or throbbing pain, redness, warmth, and sometimes bluish discoloration in the hands and feet. This can be triggered by warmth or exercise and relieved by cooling.
- Headaches: Can be persistent or migraine-like.
- Dizziness and Lightheadedness.
- Visual Disturbances: Transient blurred vision, “light shows” (ocular migraines), or temporary vision loss.
- Paresthesias: Tingling, numbness, or prickling sensations in the hands and feet.
Large Blood Vessel Issues (Macrovascular Thrombosis)
- Stroke or Transient Ischemic Attack (TIA): Sudden weakness or numbness on one side of the body, difficulty speaking, vision changes, severe headache, dizziness, loss of balance.
- Heart Attack (Myocardial Infarction): Chest pain, shortness of breath, nausea, sweating, pain radiating to the arm, jaw, or back. This is less common in ET than in other conditions but can occur.
- Deep Vein Thrombosis (DVT): Pain, swelling, redness, and warmth in a leg (usually the calf).
- Pulmonary Embolism (PE): Sudden shortness of breath, chest pain, cough (possibly with blood), rapid heartbeat. This occurs if a DVT breaks off and travels to the lungs.
- Abdominal Vein Thrombosis (e.g., Hepatic or Mesenteric Vein Thrombosis): Abdominal pain, bloating, nausea, vomiting.
Symptoms Related to Bleeding (Hemorrhage)
Paradoxically, despite having a high platelet count, some individuals with essential thrombocythemia, particularly those with extremely high counts (often >1,000,000/µL), can experience bleeding. This is because the overproduced platelets may be dysfunctional and lack the ability to effectively contribute to blood clotting.
- Easy Bruising: Bruising more easily than usual without a significant injury.
- Nosebleeds (Epistaxis): Frequent or severe nosebleeds.
- Bleeding Gums: Especially after brushing teeth.
- Gastrointestinal (GI) Bleeding: May manifest as black, tarry stools (melena) or blood in the stool (hematochezia).
- Heavy Menstrual Periods (Menorrhagia): In women.
- Less Common: Blood in the urine (hematuria).
Other General Signs and Symptoms
These are less specific to ET but can occur.
- Fatigue: Feeling unusually tired or weak.
- Enlarged Spleen (Splenomegaly): May cause discomfort or fullness in the upper left abdomen, early satiety (feeling full quickly after eating), or even pain.
- Weight Loss: Unexplained weight loss.
- Night Sweats: Excessive sweating during sleep.
- Low-Grade Fevers: Persistent low-grade temperature elevations.
- Itching (Pruritus): Can be generalized or more pronounced after a warm shower (aquagenic pruritus).
Complications of Essential Thrombocythemia (ET)
Essential Thrombocythemia (ET), while often indolent, can lead to several complications, primarily related to the increased and often dysfunctional platelets. These complications can significantly impact a patient’s morbidity and, in rare cases, mortality.
- Major Arterial Thrombosis: Leading to significant end-organ damage such as severe stroke with permanent disability, large myocardial infarction causing significant heart dysfunction, or limb ischemia requiring amputation.
- Major Venous Thrombosis with Severe Consequences: Such as a massive pulmonary embolism leading to respiratory failure or death, or Budd-Chiari syndrome causing irreversible liver damage or failure.
- Transformation to Acute Leukemia (AML): This is a rare but severe complication, affecting less than 1% of essential thrombocythemia (ET) patients per year. It involves the transformation of abnormal blood stem cells into acute leukemia, a rapidly progressing cancer.
- Transformation to Primary Myelofibrosis (PMF) with Severe Myelofibrosis: This is the most concerning complication, occurring in approximately 1% of essential thrombocythemia (ET) patients per year. Leading to significant bone marrow failure, scarring of the bone marrow, severe anemia and thrombocytopenia requiring frequent transfusions, debilitating splenomegaly causing significant discomfort and complications, and increased risk of infections and bleeding.
- Severe Pregnancy Complications Leading to Loss of Pregnancy or Maternal Morbidity: Such as stillbirth, severe preeclampsia leading to organ damage, or life-threatening thrombotic events during pregnancy or postpartum.
- Intracranial Hemorrhage: A rare but life-threatening bleeding event within the brain.
- Severe Gastrointestinal Hemorrhage Requiring Major Intervention: Leading to significant blood loss, hemodynamic instability, and potentially requiring surgery.
Laboratory Investigations and Diagnosis of Essential Thrombocythemia (ET)
The laboratory investigation of Essential Thrombocythemia (ET) is a multi-step process designed to confirm a clonal myeloproliferative state and, crucially, to exclude reactive causes and other “mimic” myeloid neoplasms (especially pre-fibrotic myelofibrosis).
Peripheral Blood Analysis (The Initial Screen)
The hallmark of ET is persistent, unexplained thrombocytosis.
- Complete Blood Count (CBC):
- Platelet Count: Must be ≥ 450 x 109/L. It should be persistent (usually measured over 3-6 months) to rule out transient reactive causes.
- Hemoglobin/Hematocrit: Typically normal. Significant elevation may point toward Polycythemia Vera (PV).
- White Blood Cell (WBC) Count: Usually normal or only mildly elevated. A high neutrophil count may suggest Pre-fibrotic Primary Myelofibrosis (pre-PMF).
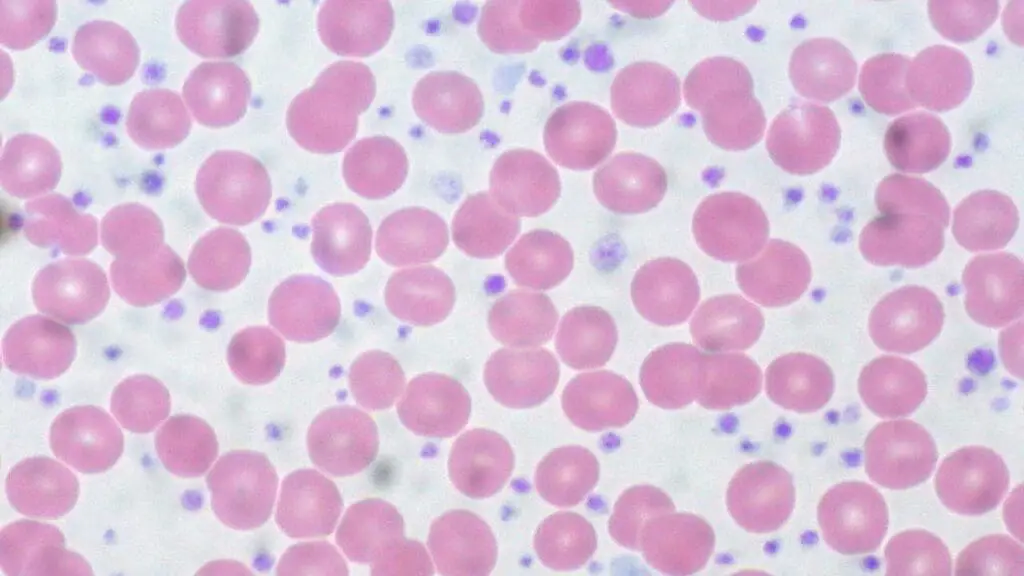
- Peripheral Blood Smear:
- Shows marked thrombocytosis with significant platelet anisocytosis (variation in size).
- Giant Platelets: Large, granular platelets are often visible.
- Absence of Leukoerythroblastosis: Unlike myelofibrosis, ET should not typically show teardrop cells (dacrocytes) or immature myeloid/erythroid cells in the periphery.
Bone Marrow Examination
A bone marrow aspiration and trephine biopsy (BMTB) is mandatory to confirm the diagnosis.
- Cellularity: Usually normocellular or only mildly hypercellular for the patient’s age.
- Megakaryocyte Morphology (The “Staghorn” Sign): There is a proliferation of large to giant megakaryocytes. They feature hyperlobulated nuclei, often described as “staghorn-like.” They are usually distributed singly or in loose groups, not in the dense, atypical clusters seen in PMF.
- Reticulin Fibrosis: Grade 0 or 1 is acceptable. Anything ≥ Grade 2 suggests progression to Post-ET Myelofibrosis or a primary diagnosis of PMF.
- Other Lineages: Erythropoiesis and granulopoiesis should appear normal, without a significant “left shift” or dysplasia.

Molecular and Genetic Testing
Molecular markers are now central to the diagnosis, providing evidence of clonality.
- Driver Mutations (The Big Three):
- JAK2 (V617F): Found in ~55–60% of cases.
- CALR (Calreticulin): Found in ~20–25% (Type 1 and Type 2).
- MPL (Thrombopoietin receptor): Found in ~3–5% (usually W515K/L).
- Triple-Negative ET: Approximately 10–15% of patients lack all three mutations. In these cases, it is vital to search for:
- Non-driver mutations (via NGS): Mutations in ASXL1, EZH2, TET2, IDH1/2, SRSF2, or SF3B1 can support the diagnosis of a clonal disorder.
- Cytogenetics: While often normal in ET, testing for the Philadelphia Chromosome (t(9;22); BCR-ABL1) is mandatory to rule out Chronic Myeloid Leukemia (CML), which can present solely with high platelets.
Ancillary and Differential Workup
Because reactive thrombocytosis is much more common than ET, laboratory investigations must rule out secondary causes:
- Iron Studies (Ferritin/Serum Iron): To rule out iron deficiency anemia, a common cause of reactive thrombocytosis. (Note: Iron deficiency can also mask Polycythemia Vera).
- Inflammatory Markers (CRP/ESR): High levels suggest reactive thrombocytosis due to infection, malignancy, or autoimmune disease.
- Lactate Dehydrogenase (LDH): Usually normal or slightly high in ET. Significantly elevated LDH is a “red flag” for pre-PMF or overt myelofibrosis.
- Uric Acid: May be elevated due to increased cell turnover.
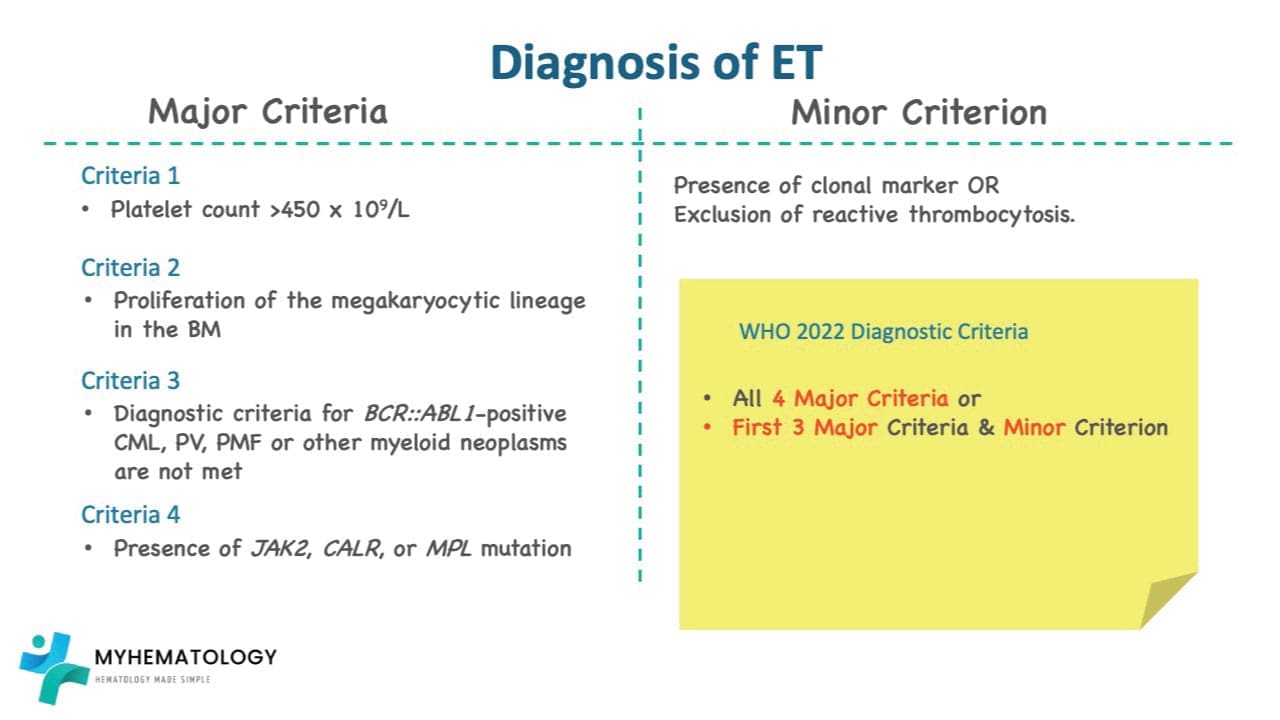
Summary Checklist for Diagnosis (WHO 2022)
To confirm ET, a patient must meet all 4 Major Criteria OR the first 3 Major and 1 Minor Criterion:
| Type | Criterion |
| Major 1 | Platelet count ≥ 450 x 109/L. |
| Major 2 | BM biopsy: Large/giant megakaryocytes with hyperlobulated nuclei; no significant granulocytic or erythroid increase; no/minor fibrosis. |
| Major 3 | Not meeting WHO criteria for BCR-ABL1+ CML, PV, PMF, MDS, or other myeloid neoplasms. |
| Major 4 | Presence of JAK2, CALR, or MPL mutation. |
| Minor | Presence of a clonal marker (e.g., NGS mutation) OR absence of evidence for reactive thrombocytosis. |
Differential Diagnosis of Thrombocytosis
Thrombocytosis, defined as an elevated platelet count, can occur due to various underlying conditions. Differentiating between primary (clonal) causes like essential thrombocythemia (ET) and secondary (reactive) causes is crucial for determining the most appropriate management approach and prognosis.
Reactive Thrombocytosis
Most common cause of thrombocytosis (80-90% of cases). Characterized by an elevated platelet count as a response to another underlying condition. Causes include:
- Inflammatory conditions: Infections, autoimmune diseases (e.g., rheumatoid arthritis, systemic lupus erythematosus)
- Tissue injury and surgery: Trauma, major burns, post-operative states
- Blood loss: Iron deficiency anemia, chronic blood loss (e.g., from peptic ulcers)
- Certain medications: Erythropoietin (EPO) therapy, corticosteroids, diuretics
- Splenomegaly (enlarged spleen) from various causes (e.g., liver cirrhosis, portal hypertension)
Primary Thrombocytosis (Clonal Myeloproliferative Neoplasms)
Less common than reactive thrombocytosis, accounting for 10-20% of cases. Characterized by clonal (abnormal) growth of blood cell precursors in the bone marrow, leading to increased platelet production. Examples include:
- Essential Thrombocythemia (ET): Discussed in detail above.
- Chronic Myelogenous Leukemia (CML): Characterized by the presence of the Philadelphia chromosome and involves other blood cell lineages besides platelets.
- Polycythemia Vera (PV): Primarily affects red blood cell production but can also present with thrombocytosis.
- Primary Myelofibrosis (PMF): Causes bone marrow scarring, often accompanied by thrombocytosis.
Spurious Thrombocytosis
This is a rare occurrence where laboratory errors can lead to an artificially high platelet count due to issues with blood collection or processing.
Pre-fibrotic Primary Myelofibrosis
Distinguishing between Essential Thrombocythemia (ET) and Pre-fibrotic Primary Myelofibrosis (pre-PMF) is one of the most significant challenges in hematopathology. While both conditions often present with an elevated platelet count (thrombocytosis) and shared driver mutations (JAK2, CALR, or MPL), their clinical trajectories and management strategies differ substantially.
The differentiation relies almost entirely on an expert histological assessment of a bone marrow trephine biopsy.
The primary distinction is found in the appearance and organization of the megakaryocytes and the overall cellularity of the marrow.
| Feature | Essential Thrombocythemia (ET) | Pre-fibrotic Myelofibrosis (pre-PMF) |
| Megakaryocytes | Large to giant; “Staghorn” nuclei (deeply lobulated); Mature cytoplasm. | Atypical; “Cloud-like” or bulbous nuclei (hypolobulated); Coarse chromatin. |
| MK Clustering | Generally loose or absent; scattered. | Frequent dense clustering or large groups. |
| Cellularity | Normocellular (age-appropriate). | Hypercellular (increased for age). |
| Granulopoiesis | Normal; not expanded. | Increased (Granulocytic proliferation). |
| Erythropoiesis | Normal; not expanded. | Often decreased or “shifted.” |
| Reticulin Fibrosis | Grade 0 (Rarely Grade 1). | Grade 0 or Grade 1 (never ≥ Grade 2). |
Additional Considerations
- Myelodysplastic Syndromes (MDS): Can sometimes present with thrombocytosis, but usually also involve other blood cell abnormalities.
- Paraneoplastic syndromes: Certain cancers can trigger secondary thrombocytosis.
- Splenomegaly (enlarged spleen): Can trap platelets, leading to a falsely elevated peripheral blood count.
- Thrombocytosis essentialia: A rare, non-malignant condition with persistent thrombocytosis without other identifiable causes.
Distinguishing between these entities requires a comprehensive evaluation including
- Detailed medical history and physical examination to identify potential underlying conditions.
- Complete blood count (CBC) to assess platelet count and other blood cell parameters.
- Bone marrow examination: May be necessary to evaluate for specific features suggestive of ET or other MPNs.
- Mutation analysis: Testing for JAK2, CALR, and MPL mutations can be helpful in differentiating ET from reactive thrombocytosis.
Essential Thrombocythemia (ET) Treatment and Management
The management of Essential Thrombocythemia (ET) differs fundamentally from the treatment of solid tumors or aggressive leukemias. Because Essential Thrombocythemia (ET) is a chronic, indolent neoplasm, the primary goal of therapy is not a “cure” (which is currently only possible with an allogeneic stem cell transplant, a procedure far too risky for standard ET), but rather the prevention of hemorrhagic and thrombotic complications and the alleviation of symptom burden, without increasing the risk of leukemic transformation.
Treatment decisions are highly individualized and driven by formal risk stratification.
Low-risk vs. High-risk Stratification
Patients are categorized into low-risk and high-risk groups based on factors like age, presence of symptoms, and cardiovascular risk factors. This helps guide treatment decisions.
IPSET-Thrombosis Model (Revised) for Thrombotic Risk in ET
| Risk Factors | Points |
| Age ≥ 60 years | 2 |
| Prior Thrombotic Event | 2 |
| JAK2 V617F Mutation | 1 |
| Cardiovascular Risk Factors (CVRFs)* | 1 |
*CVRFs include one or more of the following: hypertension, diabetes mellitus, smoking.
Risk Stratification Categories based on Total Points
| Risk Category | Total Points | Annual Thrombotic Risk | General Treatment Strategy |
| Low | 0 | 1.03% | Low-dose aspirin (75-100 mg daily) is generally recommended. Observation alone may be considered in very low-risk individuals without other risk factors, with close monitoring. |
| Intermediate | 1-2 | 2.35% | Low-dose aspirin (75-100 mg daily) is usually recommended. Cytoreductive therapy (e.g., hydroxyurea or anagrelide) may be considered, especially if platelet count is persistently high (>1000 x 10^9/L) or if other vascular risk factors are present. Individualized assessment is key. |
| High | ≥ 3 | 3.56% | Low-dose aspirin (75-100 mg daily) is generally recommended (unless contraindicated). Cytoreductive therapy (e.g., hydroxyurea, anagrelide, or interferon-alpha) is usually indicated to reduce the platelet count and thrombotic risk. |
Core Treatment Modalities
A. Antiplatelet Therapy
- Low-Dose Aspirin (LDA) (75–100 mg/day): The cornerstone for preventing microvascular symptoms (erythromelalgia, atypical ocular migraines) and arterial thrombosis. It is indicated for most patients, particularly those who are JAK2 positive.
- The aVWS Caveat: In patients with extreme thrombocytosis (platelets > 1,000–1,500 x 10⁹/L), the sheer number of platelets can consume large von Willebrand Factor (vWF) multimers, leading to acquired von Willebrand Syndrome (aVWS). In these patients, prescribing aspirin can trigger severe bleeding. A Ristocetin Cofactor assay and vWF antigen panel must be checked before initiating LDA in extreme thrombocytosis.
B. Cytoreductive Therapy
Cytoreduction aims to normalize the platelet count (typically targeting < 400 – 600 x 10⁹/L) and lower the leukocyte count, reducing overall blood viscosity and endothelial activation.
- Hydroxyurea (HU): First-line cytoreductive agent for High-Risk ET patients of advanced age. It is an antimetabolite (ribonucleotide reductase inhibitor) that impairs DNA synthesis. It is generally well-tolerated but can cause macrocytosis (MCV elevation is expected and serves as a marker of compliance), mucocutaneous ulcers, and cytopenias.
- Interferon-alpha (IFN-α) / Pegylated Interferon (Peg-IFN-α-2a / Ropeginterferon alfa-2b): First-line for younger patients (< 60 years) requiring cytoreduction, and the only safe option during pregnancy. It modulates the immune response and selectively suppresses the malignant megakaryocytic clone. Unlike HU, interferons have the potential for disease modification. Prolonged use can induce deep molecular remissions (reducing the JAK2 variant allele frequency). Ropeginterferon is administered less frequently (every 2 weeks) and is better tolerated than older, non-pegylated forms.
- Anagrelide: Second-line agent, primarily used when patients are intolerant to or refractory to HU, or used in combination with HU. It is a phosphodiesterase III (PDE3) inhibitor that specifically inhibits the maturation of megakaryocytes into platelets without significantly affecting other cell lines. Because of its PDE3 inhibition, it has positive inotropic and chronotropic effects on the heart. It is contraindicated in patients with severe cardiovascular disease due to the risk of palpitations, tachycardia, and heart failure. It also carries a slightly higher risk of progression to myelofibrosis compared to HU.
C. Systemic Anticoagulation
If a patient has suffered a venous thromboembolic event (e.g., Deep Vein Thrombosis, Pulmonary Embolism, or Splanchnic Vein Thrombosis), Low-Molecular-Weight Heparin (LMWH) or Direct Oral Anticoagulants (DOACs) are required, often in combination with cytoreduction to lower the overall thrombotic drive.
Emerging and Refractory Management
When patients fail first and second-line therapies, or suffer from severe constitutional symptoms (intractable fatigue, profound pruritus, weight loss) driven by the systemic inflammation characteristic of MPNs, targeted therapies are considered:
- JAK Inhibitors (e.g., Ruxolitinib): While primarily approved for Myelofibrosis and Polycythemia Vera, Ruxolitinib is increasingly used (often off-label or in trials) for ET patients who are HU-resistant or intolerant. It is exceptionally effective at shrinking enlarged spleens and extinguishing systemic inflammatory symptoms, though its effect on modifying the actual disease trajectory in Essential Thrombocythemia (ET) is still under investigation.
Pregnancy with Essential Thombocythemia
Managing pregnancy in a patient with Essential Thrombocythemia (ET) requires a multidisciplinary approach involving hematologists and maternal-fetal medicine (MFM) specialists. While most pregnancies in ET patients are successful, there is a significantly higher risk of first-trimester miscarriage, placental infarction, pre-eclampsia, and intrauterine growth restriction (IUGR).
Pre-conception Planning
Effective management begins before conception. The goal is to optimize the mother’s health and switch to pregnancy-safe medications.
- Discontinuation of Teratogens: Hydroxyurea and Anagrelide must be stopped at least 3–6 months before attempting to conceive due to their known teratogenic effects.
- Cardiovascular Optimization: Address modifiable risk factors such as hypertension or smoking, which can compound the thrombotic risk during pregnancy.
- Baseline Assessment: Obtain a baseline CBC, ferritin levels, and a history of previous pregnancy complications to stratify risk.
Risk Stratification in Pregnancy
Pregnancy in ET is categorized by both maternal thrombotic risk and obstetric risk.
| Risk Category | Criteria | Management Strategy |
| Low Risk | No prior thrombosis, no prior pregnancy complications, and platelets < 1,000–1,500 x 10⁹/L. | Low-dose Aspirin (LDA). |
| High Risk | Prior thrombosis OR prior major pregnancy complication (e.g., severe pre-eclampsia, stillbirth, repeated miscarriages). | LDA + Low-Molecular-Weight Heparin (LMWH) and/or Interferon. |
Pharmacological Management
- Antiplatelet Therapy: Low-Dose Aspirin (75–100 mg) is recommended for all pregnant patients with ET unless there is a contraindication (e.g., acquired von Willebrand Syndrome with very high platelets). It improves placental blood flow and reduces the risk of pre-eclampsia.
- Anticoagulation: Low-Molecular-Weight Heparin (LMWH) is often added in the postpartum period for all patients. It is used throughout pregnancy for those in the “High Risk” category to prevent placental thrombosis and venous thromboembolism (VTE).
- Cytoreductive Therapy: Interferon-alpha (IFN-α) is the only cytoreductive agent considered safe during pregnancy. It does not cross the placenta and is used if:
- Platelet counts exceed 1,000–1,500 x 10⁹/L.
- There is a history of major thrombosis or severe pregnancy complications.
- The patient is symptomatic (e.g., severe headaches, erythromelalgia).
- Pegylated Interferon (longer-acting) is often preferred for better tolerance and compliance.
Monitoring and Delivery
Management requires vigilant monitoring of both the marrow activity and the fetus.
- Hematological Monitoring: CBC should be checked monthly. Platelet counts often naturally decrease during the second and third trimesters due to hemodilution and increased consumption, which may allow for a reduction in medication.
- Obstetric Monitoring: Serial ultrasounds and Doppler studies are essential to monitor fetal growth and uterine artery resistance, identifying early signs of placental insufficiency.
- Delivery Planning: Aspirin is typically stopped 7 days before an elective induction or C-section. LMWH is usually paused 12–24 hours before delivery to allow for regional anesthesia (epidural).
Postpartum Care
The 6–12 weeks following delivery represent the highest risk period for maternal thrombosis.
- VTE Prophylaxis: LMWH should be restarted 6–12 hours after delivery and continued for at least 6 weeks.
- Resuming Previous Therapy: Patients can usually transition back to Hydroxyurea or Anagrelide postpartum, but these are generally not recommended during breastfeeding. If the mother wishes to breastfeed, Interferon remains the treatment of choice.
Prognosis
Life expectancy with proper management
The good news is that with proper management and regular follow-up, individuals with essential thrombocythemia (ET) can have a near-normal lifespan.
- Median survival for essential thrombocythemia (ET) patients is estimated to be around 18 years, which is similar to the general population of the same age and sex.
- Low-risk patients, defined by specific criteria like younger age and absence of symptoms, can have an even better prognosis with a median survival exceeding 26 years.
Importance of regular monitoring and follow-up
However, it is essential to understand that essential thrombocythemia (ET) is a chronic condition, and regular monitoring and follow-up are crucial for several reasons:
- Early detection and management: Allows for timely intervention to reduce the risk of complications like blood clots and potential progression to other conditions.
- Monitoring treatment response: Ensures the chosen treatment is effective in controlling the platelet count and managing symptoms.
- Assessing for complications: Enables healthcare professionals to identify potential complications like myelofibrosis or leukemia early, allowing for prompt initiation of appropriate treatment.
Frequently Asked Questions (FAQs)
Is essential thrombocythemia serious?
While essential thrombocythemia (ET) itself is a chronic condition and technically classified as a cancer, it is generally not considered a serious illness in the same way as other cancers, provided it is properly managed. With proper diagnosis, treatment, and regular follow-up, individuals with essential thrombocythemia (ET) can have a near-normal lifespan. The median survival for essential thrombocythemia (ET) patients is estimated to be around 18 years, similar to the general population of the same age and sex.
Can essential thrombocytosis be cured?
Unfortunately, essential thrombocythemia (ET) cannot be cured. It is a chronic condition caused by genetic mutations that lead to abnormal blood cell production. While treatments can effectively manage the symptoms and significantly reduce the risk of complications, they cannot permanently eliminate the underlying cause.
Is essential thrombocythemia a form of leukemia?
While Essential Thrombocythemia (ET) is a type of blood cancer classified as a myeloproliferative neoplasm (MPN), it is not a form of leukemia. Leukemia is a broader term for cancers that originate in the blood-forming cells of the bone marrow and typically involve an overproduction of abnormal white blood cells. MPNs, on the other hand, are characterized by the overproduction of one or more blood cell types (red blood cells, white blood cells, or platelets). In ET, the primary issue is the overproduction of platelets.
Why does essential thrombocythemia cause bleeding?
Essential Thrombocythemia (ET) can paradoxically cause bleeding despite a high platelet count due to qualitative defects in the function of the overproduced platelets. While there are a large number of platelets, they may not be working properly.
- Medication Effects: It’s also important to consider that treatments for ET, such as aspirin, are antiplatelet agents and can increase the risk of bleeding.
- Abnormal Platelet Morphology and Granule Content: The rapidly proliferating megakaryocytes in ET often produce platelets that are structurally and biochemically abnormal. These platelets may have altered size, shape, and a deficiency or dysfunction in their granules. Granules contain essential substances (like ADP, thromboxane A2, and von Willebrand factor) that are crucial for platelet activation, adhesion, and aggregation – the key steps in forming a stable blood clot.
- Impaired Platelet Activation and Aggregation: Due to the abnormalities, the platelets in ET may not be as responsive to activating signals or may not aggregate effectively to form a proper platelet plug at the site of injury. This can lead to prolonged bleeding times and increased susceptibility to hemorrhage.
- Acquired von Willebrand Factor (vWF) Deficiency (Rare): In some cases of very high platelet counts, there can be an acquired deficiency of von Willebrand factor. vWF is a protein that helps platelets adhere to damaged blood vessel walls. The high number of platelets can bind to and consume vWF, leading to reduced levels and impaired platelet adhesion.
- Interaction with the Vessel Wall: The abnormal platelets might also have altered interactions with the endothelial cells lining the blood vessels, potentially contributing to a fragile vascular environment that is more prone to bleeding.
Why does essential thrombocythemia cause fatigue?
Fatigue in Essential Thrombocythemia is a common and often significant symptom likely arising from a combination of factors related to the chronic myeloproliferative nature of the disease, including the body’s response to abnormal blood cell production, the release of inflammatory cytokines, potential microcirculatory disturbances affecting oxygen delivery, and in some cases, mild anemia, splenomegaly, psychological distress associated with a chronic condition, and side effects of medications used for treatment.
Is essential thrombocythemia hereditary?
In most cases, essential thrombocythemia (ET) is not considered hereditary. It arises from acquired genetic mutations that occur after conception in the early blood-forming cells. These mutations are not inherited from parents and are not passed onto offspring.
However, there are rare cases where essential thrombocythemia (ET) can have a familial component, meaning it can occur in multiple family members. This is known as familial essential thrombocythemia and is estimated to account for approximately 10% of all essential thrombocythemia (ET) cases.
- Non-hereditary (Sporadic) essential thrombocythemia (ET): This is the most common form, accounting for 90% of cases. Mutations occur in the bone marrow after conception, not inherited from parents. There is no increased risk of passing the condition to offspring.
- Familial Essential Thrombocythemia: This occurs in less than 10% of cases. It is caused by germline mutations (mutations present in the egg or sperm) that are inherited from a parent. This increases the risk of developing essential thrombocythemia (ET) for offspring of the affected individual. However, having a family history of essential thrombocythemia (ET) does not guarantee that offspring will develop it, and the risk is still relatively low.
Genetic testing can help identify individuals with a familial predisposition to essential thrombocythemia (ET). However, this test is not routinely performed for everyone with essential thrombocythemia (ET).
Does essential thrombocythemia get worse over time?
While not guaranteed, some factors can increase the risk of complications and potentially lead to a more progressive course.
- Advanced age: The risk of complications generally increases with age.
- High platelet count: A very high platelet count (above 1 million per microliter) is associated with a higher risk of complications.
- Specific mutations: The presence of certain gene mutations, such as TP53 mutation, can be associated with a higher risk of transformation to AML.
Can you donate blood if you have essential thrombocythemia?
Individuals with essential thrombocythemia (ET) are generally not eligible to donate blood due to safety concerns. While the high platelet count may seem like a benefit for donation, it can pose potential risks. Firstly, the increased platelet count in essential thrombocythemia (ET) often involves abnormal platelet function, reducing their effectiveness in clotting. Secondly, removing a significant amount of platelets during donation could further decrease the individual’s platelet count, potentially increasing their own risk of bleeding complications. Therefore, to ensure both donor and recipient safety, individuals with essential thrombocythemia (ET) are advised not to donate blood.
What is the difference between Essential Thrombocythemia and Polycythemia Vera?
While both are myeloproliferative neoplasms, Polycythemia Vera (PV) is primarily characterized by an overproduction of red blood cells (often accompanied by elevated white cells and platelets) and is almost exclusively driven by JAK2 mutations. ET solely involves an overproduction of platelets and can be driven by JAK2, CALR, or MPL mutations.
Can a patient with ET have a safe pregnancy?
Yes, but it is considered a high-risk pregnancy requiring coordination between a hematologist and a maternal-fetal medicine specialist. Teratogenic medications like Hydroxyurea must be discontinued. Management typically relies on low-dose aspirin, low-molecular-weight heparin (LMWH), and occasionally Pegylated Interferon to control counts and prevent placental thrombosis.
What happens if a patient becomes resistant to Hydroxyurea?
If a patient experiences an inadequate response, uncontrolled counts, or intolerable cytopenias/side effects from Hydroxyurea, second-line therapies are deployed. These include Anagrelide, Pegylated Interferon, or targeted clinical trial options.
Are there specific dietary or lifestyle recommendations for ET?
While no diet cures ET, an anti-inflammatory diet (such as the Mediterranean diet) is frequently recommended to mitigate the chronic systemic inflammation associated with MPNs. Strict management of cardiovascular risk factors including smoking cessation, blood pressure control, and lipid management is absolutely critical to lowering the compounded risk of thrombosis.
How do hematopathologists distinguish between ET and pre-fibrotic Myelofibrosis?
Differentiation relies heavily on expert morphological evaluation of a bone marrow biopsy. Pre-PMF presents with highly atypical, abnormally maturing megakaryocytes and increased overall bone marrow cellularity for the patient’s age. True ET features large, fully mature megakaryocytes without significant cellular atypia or early reticulin fibrosis.
Glossary of Related Medical Terms
- Cytoreductive Therapy: Medical treatments, such as Hydroxyurea, designed to suppress bone marrow activity and lower the number of circulating blood cells.
- Erythromelalgia: A rare neurovascular complication characterized by episodic burning pain, warmth, and intense redness in the extremities, often driven by microvascular platelet thrombosis in MPNs.
- Megakaryocyte: A large, multinucleated bone marrow cell responsible for the production and release of blood thrombocytes (platelets).
- Myeloproliferative Neoplasm (MPN): A group of blood cancers characterized by the clonal overproduction of one or more types of mature blood cells in the bone marrow.
- Pre-fibrotic Primary Myelofibrosis (pre-PMF): An early, hypercellular stage of myelofibrosis that mimics ET clinically but features distinct morphological bone marrow atypia and carries a higher risk of disease progression.
- Reticulin Stain: A specialized histological stain used on bone marrow biopsies to detect and grade the network of reticulin fibers, essential for ruling out myelofibrosis during an ET diagnosis.
- Thrombopoietin (TPO): A glycoprotein hormone produced primarily by the liver that regulates the proliferation and maturation of megakaryocytes.
Disclaimer: This article is intended for informational purposes only and is specifically targeted towards medical students. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- https://mpn-hub.com/medical-information/international-consensus-classification-2022-for-myeloproliferative-neoplasms
- Gianelli U, Thiele J, Orazi A, Gangat N, Vannucchi AM, Tefferi A, Kvasnicka HM. International Consensus Classification of myeloid and lymphoid neoplasms: myeloproliferative neoplasms. Virchows Arch. 2023 Jan;482(1):53-68. doi: 10.1007/s00428-022-03480-8. Epub 2022 Dec 29. PMID: 36580136; PMCID: PMC9852206.
- Goldberg S, Hoffman J. Clinical Hematology Made Ridiculously Simple, 1st Edition: An Incredibly Easy Way to Learn for Medical, Nursing, PA Students, and General Practitioners (MedMaster Medical Books). 2021.
- Keohane EM, Otto CN, Walenga JM. Rodak’s Hematology 6th Edition (Saunders). 2019.
- https://www.uptodate.com/contents/essential-thrombocythemia-treatment-and-prognosis
- Kaur A, Gohari P. Essential Thrombocytosis. [Updated 2025 Aug 9]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2026 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK539709/
- Tefferi A, Vannucchi AM, Barbui T. Essential thrombocythemia: 2024 update on diagnosis, risk stratification, and management. Am J Hematol. 2024;99(4):697-718. doi:10.1002/ajh.27216
- Godfrey, A. L., Green, A. C., & Harrison, C. N. (2023). Essential thrombocythemia: challenges in clinical practice and future prospects. Blood, 141(16), 1943–1953. https://doi.org/10.1182/blood.2022017625
- Guglielmelli, P., Szuber, N., Gangat, N., Capecchi, G., Maccari, C., Harnois, M., Karrar, O., Abdelmagid, M., Balliu, M., Nacca, E., Atanasio, A., Sestini, I., Désilets, A., Loscocco, G. G., Rotunno, G., Busque, L., Tefferi, A., & Vannucchi, A. M. (2024). CALR mutation burden in essential thrombocythemia and disease outcome. Blood, 143(13), 1310–1314. https://doi.org/10.1182/blood.2023023428
- Tefferi, A., & Barbui, T. (2024). Aspirin use in essential thrombocythemia: Once-daily or twice-daily or not at all?. American journal of hematology, 99(8), 1450–1453. https://doi.org/10.1002/ajh.27369
- Brum, I. S. D. C., Goncalves, J., Zanchetta, M., Xerém, B., Lanziani, R., Haiut, M., Hada, M., Apa, A. G., & Cordovil, K. (2024). Essential thrombocythemia: nutritional management in weight loss and malnutrition. The Pan African medical journal, 47, 93. https://doi.org/10.11604/pamj.2024.47.93.32594
- Picard, A., Bayart, S., Deparis, M., De Maricourt, C. D., Haro, S., Jourdain, A., Mallebranche, C., Rialland, F., Luque Paz, D., Pastoret, C., Gandemer, V., & Cousin, E. (2025). Polycythemia vera and essential thrombocythemia in children, still a challenge for pediatricians. European journal of pediatrics, 184(2), 173. https://doi.org/10.1007/s00431-025-05993-1
- Grenier, J. M. P., El Nemer, W., & De Grandis, M. (2024). Red Blood Cell Contribution to Thrombosis in Polycythemia Vera and Essential Thrombocythemia. International journal of molecular sciences, 25(3), 1417. https://doi.org/10.3390/ijms25031417