Key Takeaways
Chronic myeloid leukemia is a slow-growing blood cancer caused by the BCR-ABL1 fusion gene on the Philadelphia chromosome, formed by a swap between chromosomes 9 and 22.
Signs and symptoms ▾: Most patients are diagnosed in chronic phase, often by an unexpectedly high white cell count on a routine blood test, with basophilia and an enlarged spleen as classic clues.
Laboratory investigations ▾: Diagnosis is confirmed by detecting the Philadelphia chromosome (karyotyping or FISH) or the BCR-ABL1 fusion gene (quantitative PCR), and qPCR is also the gold standard for monitoring response.
Treatment and management ▾: Tyrosine kinase inhibitors are the cornerstone of chronic myeloid leukemia treatment; imatinib remains a strong first-line choice, and asciminib was approved in October 2024 as a new frontline option.
*Click ▾ for more information
Chronic myeloid leukemia (CML) is one of the great success stories of modern cancer medicine. Thirty years ago it was almost uniformly fatal. Today, most newly diagnosed patients can expect a life expectancy close to that of the general population [1,4]. The reason for that change is a single targeted drug class — the tyrosine kinase inhibitors — that shut down the very genetic accident that causes the disease.
What is chronic myeloid leukemia?
Chronic myeloid leukemia also known as CML is a slow-growing cancer of the blood and bone marrow. It begins in a single bone marrow stem cell that picks up a specific genetic change and starts dividing without stopping. The descendants of that cell flood the marrow and bloodstream, mostly as granulocytes — the family of white blood cells that includes neutrophils, eosinophils, and basophils.
Hematologists classify CML as a myeloproliferative neoplasm, meaning a bone marrow cancer that overproduces one or more blood cell types. Unlike acute leukemias, which strike fast and produce mostly immature "blast" cells, chronic-phase CML produces a full range of granulocytes at every stage of maturation. This is why it tends to creep up slowly and often discovered by accident on a routine blood test [4,7].
Who gets it?
Chronic myeloid leukemia accounts for roughly 15% of adult leukemias and is uncommon overall, with about 1 to 2 cases per 100,000 adults per year [7]. It can appear at any age but is mostly a disease of older adults; the median age at diagnosis is 60 to 65, and men are affected slightly more often than women.
What causes chronic myeloid leukemia?
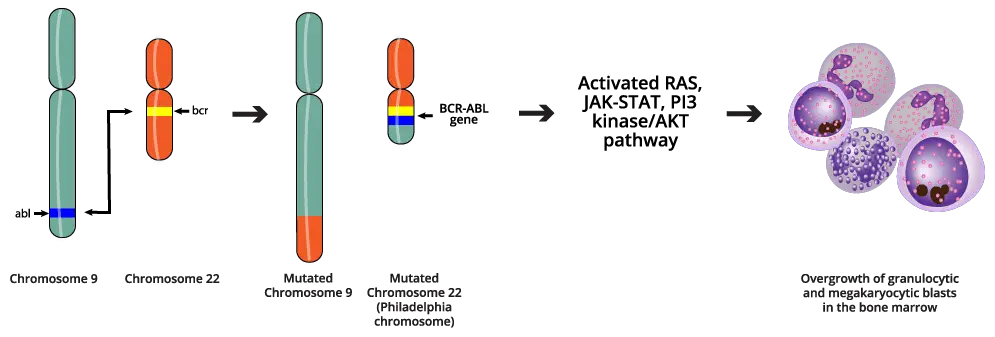
Almost every case of CML comes down to a single genetic event: the formation of an abnormal fusion gene called BCR-ABL1.
The Philadelphia (Ph) Chromosome
In a healthy cell, chromosome 9 carries a gene called ABL1, and chromosome 22 carries a gene called BCR. In CML, pieces of these two chromosomes swap places. This is called a reciprocal translocation, written as t(9;22)(q34;q11). The shortened chromosome 22 that results in the Philadelphia (Ph) chromosome, after the city where it was first identified, carries a brand-new fusion gene: BCR-ABL1 [4,7].
Why the fusion gene matters
The normal ABL1 gene makes a tyrosine kinase. A tyrosine kinase is an enzyme that switches other proteins on by adding a phosphate tag, and in healthy cells its activity is tightly controlled.
The BCR portion changes everything. It forces the new BCR-ABL1 protein to clump together with copies of itself, and that clumping locks the kinase into a permanent "on" state. This is what biologists call constitutive activity — the enzyme keeps signaling whether or not the cell needs it to.
A chronically switched-on kinase rewires the cell. It activates major growth pathways (RAS/MAPK, PI3K/AKT, JAK/STAT), blocks the normal "self-destruct" program that removes faulty cells (apoptosis), and loosens the grip that holds immature cells inside the bone marrow [4,7]. The result: too many myeloid cells, living too long, escaping into the bloodstream too early.
From chronic disease to crisis
Over time, BCR-ABL1 also breaks the cell's DNA repair machinery, allowing additional mutations to pile on. This is how the disease drifts from the controllable chronic phase into the more dangerous accelerated and blast phases if left untreated.
Clinical presentation & the 3 phases of CML
Chronic myeloid leukemia is staged in three phases. Modern TKI therapy aims to keep patients permanently in the first one [1].
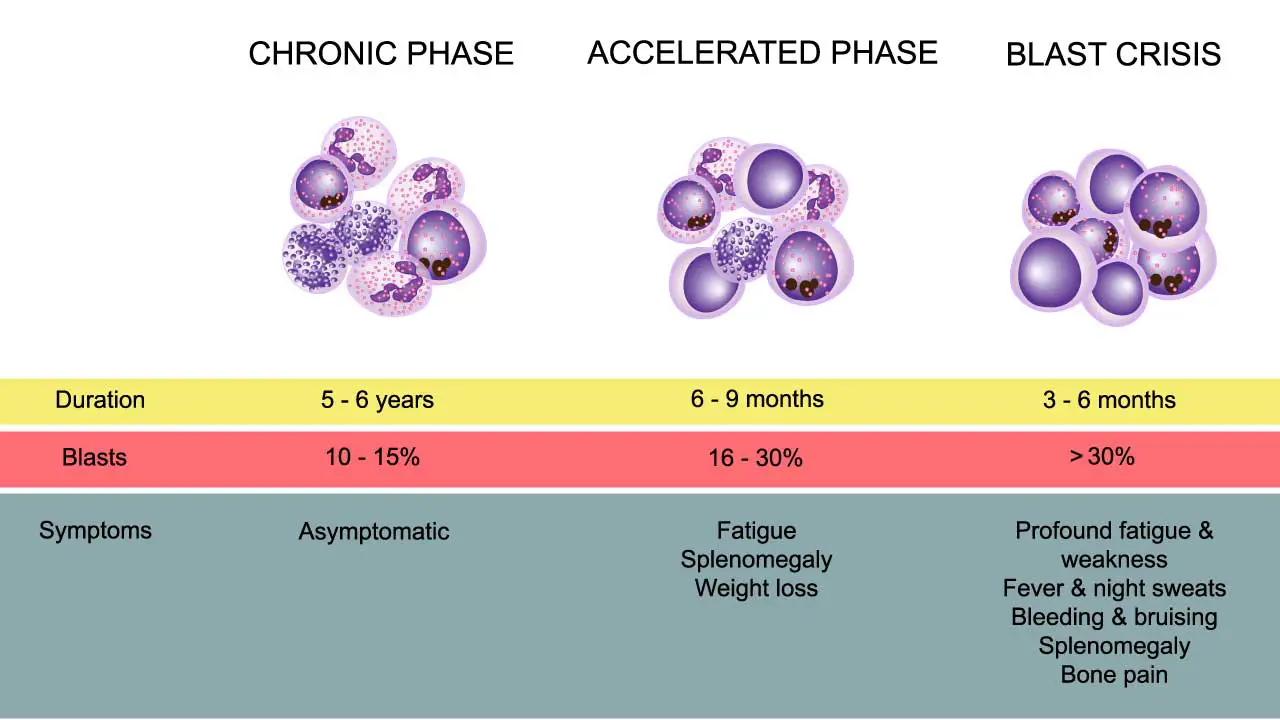
Chronic phase
About 85–90% of patients are diagnosed in this phase, and many have no symptoms at all. The disease is often picked up on a routine complete blood count.
When symptoms do appear, they tend to be vague: fatigue, low-grade fever, night sweats, and unintentional weight loss, all driven by the high turnover of leukemia cells. Patients may notice abdominal fullness or discomfort because the spleen which mops up extra blood cells becomes enlarged. Some develop bruising or nosebleeds from platelets that look normal but do not function properly. Joint pain or gout flares can occur because broken-down leukemia cells flood the body with uric acid.
On examination, the most consistent finding is splenomegaly (an enlarged spleen) below the costal margin. The liver may also be enlarged. In rare cases with very high white cell counts (≥200 × 10⁹/L), blood becomes thick enough to cause "hyperviscosity" symptoms like visual disturbances, headaches, or priapism which are a medical emergency.
Accelerated phase
The accelerated phase is a transitional warning zone where the leukemic clone starts becoming harder to control. Definitions vary slightly between WHO and ELN, but the general features include:
- 10–19% blasts in the blood or bone marrow
- Persistent low platelet counts (≤100 × 10⁹/L) or persistent high counts (thrombocytosis) that do not respond to treatment
- Rising white cell counts despite therapy
- Basophils ≥20% in the peripheral blood
- New chromosomal abnormalities appearing alongside the Philadelphia chromosome (for example, trisomy 8 or isochromosome 17q)
- Soft-tissue tumors of leukemia cells (granulocytic sarcomas)
Blast crisis
Blast crisis is CML's transformation into an acute leukemia, and it carries a poor prognosis. It is defined by ≥20% blasts in the blood or bone marrow, or by tumors of blasts outside the marrow. About 60–70% of cases transform along the myeloid line (becoming acute myeloid leukemia), and 20–30% along the lymphoid line (becoming acute lymphoblastic leukemia, which sometimes responds better to ALL chemotherapy combined with a TKI). Patients in blast crisis usually present like acute leukemia patients — with infection, bleeding, and severe cytopenias — and need urgent, intensive treatment.
Biphasic model of CML
The 5th edition of the WHO Classification (WHO-HAEM5) eliminated the Accelerated Phase (AP) as a distinct category, the International Consensus Classification (ICC) 2022 maintained it [8,9]. WHO have shifted to a biphasic model, eliminating the "accelerated phase" because modern TKIs have largely rendered it obsolete by aiming to keep patients permanently in the first phase.
Chronic phase (CP)
The vast majority of patients are diagnosed in this phase. The disease is often entirely asymptomatic and picked up on a routine complete blood count. When symptoms occur, they tend to be vague: fatigue, low-grade fever, night sweats, and unintentional weight loss, all driven by the high turnover of leukemia cells. An enlarged spleen (splenomegaly) is the most consistent physical finding, causing abdominal fullness. Features previously used to define the accelerated phase (such as rising blast counts between 10-19%, persistent low platelets, or new chromosomal abnormalities) are now classified as "high-risk features" or "warning signs" within the chronic phase, signaling that a change in TKI therapy may be required [8].
Blast phase (BP)
Blast phase (formerly blast crisis) is CML's transformation into a highly aggressive acute leukemia, carrying a poor prognosis. It is defined by 20% or more blasts in the blood or bone marrow, or by the presence of leukemic tumors outside the marrow [8]. About 60–70% of cases transform into acute myeloid leukemia (AML), and 20–30% into acute lymphoblastic leukemia (ALL). Patients in blast phase require urgent, intensive treatment, often combining acute leukemia chemotherapy with a potent TKI.
How is chronic myeloid leukemia (CML) investigated?
Diagnosis combines routine hematology with molecular and chromosomal testing.
Step 1: Blood count and blood smear
The complete blood count and peripheral blood smear usually raise the suspicion. Typical findings include:
- Leukocytosis (high white cell count), often in the range of 25 to 500 × 10⁹/L
- A full spectrum of myeloid maturation on the smear — myeloblasts, promyelocytes, myelocytes, metamyelocytes, bands, and segmented neutrophils all visible together
- Basophilia — an increase in basophils, which is highly characteristic of CML
- Eosinophilia
- Normal or elevated platelet counts in chronic phase
- A low Leukocyte Alkaline Phosphatase (LAP) score, which helps distinguish CML from a leukemoid reaction caused by infection, where LAP is high
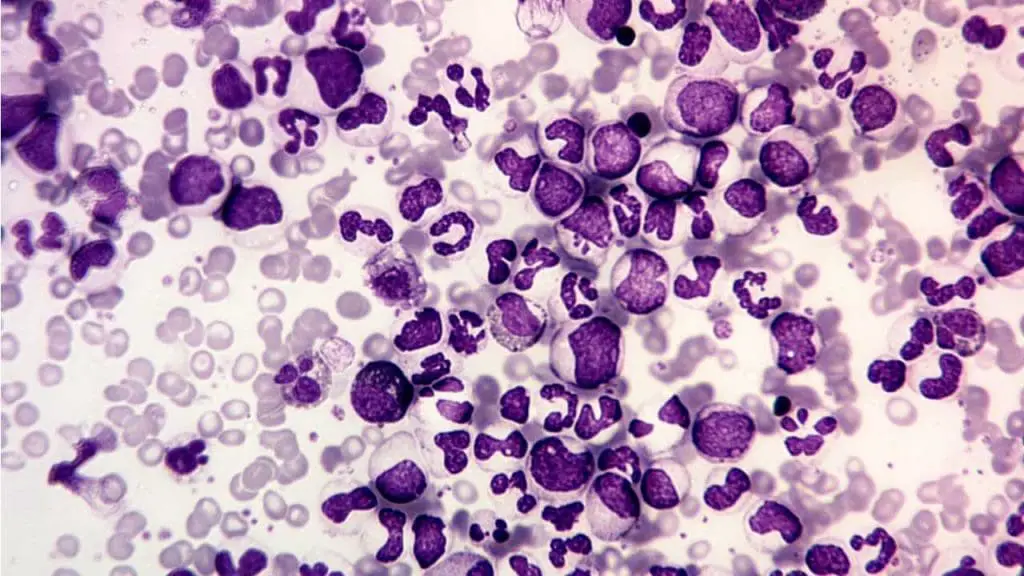
Step 2: Bone marrow examination
Bone marrow aspiration and biopsy show a hypercellular marrow (more than 80% cellularity) packed with granulocytes. The myeloid-to-erythroid ratio jumps from a normal 2:1–4:1 to 10:1–30:1. Small abnormal megakaryocytes are often increased, and reticulin fibrosis may be present, which can hint at a poorer prognosis. The blast percentage measured here defines the disease phase: <10% in chronic phase, 10–19% in accelerated phase, and ≥20% in blast crisis.
Step 3: Molecular and cytogenetic confirmation
This step is essential. CML is defined by the BCR-ABL1 fusion, and treatment is built around it.
Conventional karyotyping looks at all 46 chromosomes from a bone marrow sample. It detects the Philadelphia chromosome itself and any extra abnormalities that signal clonal evolution.
Fluorescence in situ hybridization (FISH) uses fluorescently labeled DNA probes that bind to BCR and ABL1. When the fusion is present, the two color signals merge. FISH is fast, sensitive, and works on peripheral blood. It is useful when bone marrow culture fails.
Quantitative PCR (qPCR) is the gold standard for monitoring. It measures BCR-ABL1 transcripts on the International Scale (IS), allowing labs around the world to report results in directly comparable terms. Major Molecular Response (MMR) defined as BCR-ABL1 ≤0.1% IS is a key therapeutic milestone [1,5].
While qPCR is the standard for routine monitoring, modern CML management increasingly relies on two advanced technologies. Next-Generation Sequencing (NGS) has largely replaced traditional Sanger sequencing for mutational analysis when a patient fails to meet clinical milestones, as it is highly sensitive at detecting low-burden resistance mutations before a full clinical relapse occurs [10].
Additionally, Digital Droplet PCR (ddPCR) is emerging in specialized centers as an ultra-sensitive tool for patients attempting Treatment-Free Remission (TFR), as it provides absolute quantification of the BCR-ABL1 transcript without needing a standard curve, making it ideal for detecting deep molecular responses (MR4.5 or MR5.0) [11].
Predicting prognosis: risk stratification
Before starting treatment, doctors estimate how aggressive a patient's CML is likely to be. This guides the choice of first-line TKI [1].
The Sokal score, developed before TKIs existed, uses age, spleen size, platelet count, and blood blast percentage. It is still occasionally cited but has largely been replaced.
The EUTOS Long-Term Survival (ELTS) score is the modern preference. It uses the same four variables but reweights them to reflect TKI-era outcomes, giving more weight to age. The ELTS score is designed specifically to predict leukemia-specific death—the most relevant metric today, given that most CML patients now die of non-CML related causes [12]. Patients are sorted into low, intermediate, and high-risk groups to predict long-term survival and treatment failure. ELN 2020 recommends using ELTS at diagnosis [1].
The practical implication: low- and intermediate-risk patients usually do very well on imatinib, while high-risk patients often benefit from starting on a more potent agent.
Treatment and management for CML
Modern care of chronic myeloid leukemia is built on three ideas: switch off BCR-ABL1, monitor the response carefully, and aim for the deepest possible remission so that some patients can eventually stop treatment. The goal is deep and sustained molecular remission. A deep dive into the current treatment and management of CML can be found in another article titled 'Chronic Myeloid Leukemia Treatment Strategies'.
Tyrosine kinase inhibitors (TKIs)
Tyrosine kinase inhibitors (TKIs) work by sliding into the BCR-ABL1 protein and blocking the spot where it would normally pick up energy (the ATP-binding pocket). With BCR-ABL1 silenced, the leukemic cells lose their growth advantage [4,7].
First-in-class, well tolerated, generic, cost-effective. Endorsed by ELN 2020 as a strong first-line choice, especially in low-risk patients [1].
More potent than imatinib; faster and deeper responses. Used first-line in higher-risk patients or after imatinib failure or intolerance.
Active against most resistance mutations. Ponatinib uniquely overcomes the T315I mutation but carries a meaningful cardiovascular risk.
Binds a different pocket on the ABL protein (the myristoyl pocket) rather than the ATP-binding site. Approved by the FDA in October 2024 as a frontline option for newly diagnosed chronic-phase Ph+ CML, after the ASC4FIRST trial showed a 48-week MMR rate of about 68% versus 49% with other TKIs [2,3].
Cardiovascular Risk and TKI Selection
Because patients may be on TKIs for decades, modern CML management requires a strict cardio-oncology approach [13]. Baseline cardiovascular risk assessments including lipid profiles, blood pressure monitoring, and diabetes screening are now mandatory before selecting a TKI. A patient's baseline vascular comorbidities often dictate the choice of drug just as much as their ELTS risk score, particularly avoiding ponatinib or nilotinib in patients with a history of arterial disease or severe metabolic syndrome [1].
Side effects: what students and caregivers should know
TKIs are generally well tolerated, but side effects matter because patients usually take them for years. Common across the class are low blood counts, fatigue, nausea, diarrhea, muscle and joint pain, and skin rash. Beyond that, each drug has its own profile: nilotinib raises blood sugar and increases cardiovascular events, dasatinib can cause pleural effusions (fluid around the lungs), and ponatinib carries a meaningful risk of arterial blood clots. Asciminib's tolerability profile compared favorably with the other TKIs in the ASC4FIRST trial [3]. Most side effects can be managed with dose adjustments, supportive care, or switching agents and adherence is itself a major predictor of outcome.
Resistance and the T315I mutation
When TKIs stop working, the most common reason is a point mutation in the ABL1 kinase domain that prevents the drug from binding. Mutational analysis is recommended at confirmed treatment failure [1,5]. The most notorious mutation, T315I, blocks all TKIs except ponatinib and asciminib (which binds a different pocket entirely).
Treatment-free remission
Treatment-free remission (TFR) is now a realistic goal for some patients. Eligibility typically requires a minimum of 3 years of total TKI therapy with at least 2 years of a sustained, deep molecular response (MR4.0 or deeper), no history of advanced disease phases, and consistent prior treatment [1]. Patients who stop TKIs are monitored monthly with qPCR for the first 6 to 12 months. About 40–60% of carefully selected patients maintain TFR long-term [6]. If the disease comes back, restarting the original TKI almost always re-establishes molecular response.
A note on pregnancy
TKIs cross the placenta and can harm a developing fetus, so they are withheld during pregnancy. Patients are managed with close monitoring, sometimes interferon-alpha if treatment is essential, and the TKI is restarted after delivery [1].
Allogeneic stem cell transplant
Before TKIs, an allogeneic stem cell transplant from a donor was the only curative option. It is still potentially curative, but the risks are high. Today it is reserved for patients who fail multiple TKIs (including ponatinib), patients in accelerated phase or blast crisis, and those with mutations that resist all available drugs.
Frequently Asked Questions (FAQs)
What is chronic myeloid leukemia in simple terms?
Chronic myeloid leukemia is a slow-growing cancer of the blood and bone marrow in which the body makes too many white blood cells. It is caused by a single genetic accident that creates an abnormal gene called BCR-ABL1, which tells stem cells to keep dividing without stopping. With modern targeted drugs called tyrosine kinase inhibitors, most patients now live near-normal lifespans.
How is chronic myeloid leukemia diagnosed?
Doctors diagnose CML by combining a blood test, a bone marrow examination, and a genetic test. A complete blood count usually shows a very high white cell count with a mix of mature and immature granulocytes, plus increased basophils. The diagnosis is confirmed by detecting the Philadelphia chromosome (using karyotyping or FISH) or the BCR-ABL1 fusion gene (using quantitative PCR), which is also used to monitor treatment.
What is the first-line treatment for chronic myeloid leukemia?
The first-line treatment is a tyrosine kinase inhibitor (TKI). Imatinib is the long-standing first-line choice and remains effective and affordable. Second-generation TKIs — dasatinib, nilotinib, and bosutinib — are also approved frontline. In October 2024, the FDA approved asciminib as a new frontline option for newly diagnosed chronic-phase CML based on the ASC4FIRST trial. Choice depends on age, risk score, comorbidities, and side effects.
Can chronic myeloid leukemia be cured?
Strictly speaking, CML is not yet considered curable, but for most patients it is now controllable as a chronic disease. With consistent TKI therapy, life expectancy in chronic-phase CML is now close to that of the general population. About 40–60% of carefully selected patients who reach a deep, sustained response can stop their TKI under close monitoring (treatment-free remission). Allogeneic stem cell transplant is the only curative option but is now reserved for patients who fail multiple TKIs or progress to advanced disease.
Is chronic myeloid leukemia hereditary?
No. The Philadelphia chromosome that causes CML is acquired during a person's lifetime in a single bone marrow stem cell. It is not passed from parent to child, and family members of a patient with CML are not at increased risk. Ionizing radiation is the only firmly established environmental risk factor.
What are the common side effects of TKIs in chronic myeloid leukemia?
Common side effects include low blood counts, fatigue, nausea, diarrhea, muscle and joint pain, and skin rash. Each drug has its own profile: nilotinib raises blood sugar and cardiovascular risk, dasatinib can cause fluid around the lungs, and ponatinib carries a higher risk of arterial blood clots. Side effects are usually managed with dose adjustments, supportive care, or switching to a different TKI.
Glossary of Related Medical Terms
- Allogeneic stem cell transplant — A procedure that replaces a patient's diseased bone marrow with healthy stem cells from a donor.
- Apoptosis — Programmed cell death; the body's normal way of removing old or damaged cells.
- BCR-ABL1 — The abnormal fusion gene that causes CML, formed when pieces of chromosomes 9 and 22 swap places.
- Blast — An immature blood cell. A high blast count means the bone marrow is making too many young cells that have not matured properly.
- Clonal disorder — A disease where all the abnormal cells come from one original "parent" cell that started dividing out of control.
- Constitutive activity — Stuck in the "on" position. A constitutively active enzyme keeps working even when no signal is telling it to.
- Cytogenetics — A laboratory analysis of chromosomes, used to spot abnormalities like the Philadelphia chromosome.
- Granulocytes — A family of white blood cells (neutrophils, eosinophils, basophils) that fight infection.
- Hematopoietic stem cell — A bone marrow cell that gives rise to every type of blood cell.
- Karyotyping — Looking at all 46 chromosomes under a microscope to check their structure and number.
- Major Molecular Response (MMR) — A measurable treatment goal: BCR-ABL1 transcripts at or below 0.1% on the International Scale.
- Myeloid lineage — The branch of blood cell development that produces red cells, platelets, and granulocytes (as opposed to the lymphoid lineage, which makes lymphocytes).
- Myeloproliferative neoplasm (MPN) — A group of bone marrow cancers in which the body makes too many of one or more blood cell types.
- Philadelphia chromosome — The shortened chromosome 22 that results from the t(9;22) translocation; the genetic signature of CML.
- qPCR (quantitative PCR) — A sensitive lab test that measures how much BCR-ABL1 a patient has, used to monitor treatment.
- Splenomegaly — An enlarged spleen.
- Translocation — When a piece of one chromosome breaks off and attaches to another chromosome.
- Tyrosine kinase — A type of enzyme that adds phosphate groups to other proteins, switching them on. The BCR-ABL1 protein is a faulty tyrosine kinase that never switches off.
- Tyrosine kinase inhibitor (TKI) — A drug that blocks the BCR-ABL1 enzyme; the cornerstone of modern CML treatment.
Disclaimer: This article is intended for educational and informational purposes only. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Hochhaus, A., Baccarani, M., Silver, R. T., Schiffer, C., Apperley, J. F., Cervantes, F., Clark, R. E., Cortes, J. E., Deininger, M. W., Guilhot, F., Hjorth-Hansen, H., Hughes, T. P., Janssen, J. J. W. M., Kantarjian, H. M., Kim, D. W., Larson, R. A., Lipton, J. H., Mahon, F. X., Mayer, J., Nicolini, F., … Hehlmann, R. (2020). European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia, 34(4), 966–984. https://doi.org/10.1038/s41375-020-0776-2
- Jabbour, E., & Kantarjian, H. (2024). Chronic myeloid leukemia: 2025 update on diagnosis, therapy, and monitoring. American journal of hematology, 99(11), 2191–2212. https://doi.org/10.1002/ajh.27443
- Hochhaus, A., Wang, J., Kim, D. W., Kim, D. D. H., Mayer, J., Goh, Y. T., le Coutre, P., Takahashi, N., Kim, I., Etienne, G., Andorsky, D., Issa, G. C., Larson, R. A., Bombaci, F., Kapoor, S., McCulloch, T., Malek, K., Yau, L., Ifrah, S., Hoch, M., … ASC4FIRST Investigators (2024). Asciminib in Newly Diagnosed Chronic Myeloid Leukemia. The New England journal of medicine, 391(10), 885–898. https://doi.org/10.1056/NEJMoa2400858
- Jabbour, E., & Kantarjian, H. (2025). Chronic Myeloid Leukemia: A Review. JAMA, 333(18), 1618–1629. https://doi.org/10.1001/jama.2025.0220
- Cross, N. C. P., Ernst, T., Branford, S., Cayuela, J. M., Deininger, M., Fabarius, A., Kim, D. D. H., Machova Polakova, K., Radich, J. P., Hehlmann, R., Hochhaus, A., Apperley, J. F., & Soverini, S. (2023). European LeukemiaNet laboratory recommendations for the diagnosis and management of chronic myeloid leukemia. Leukemia, 37(11), 2150–2167. https://doi.org/10.1038/s41375-023-02048-y
- Saussele, S., Richter, J., Guilhot, J., Gruber, F. X., Hjorth-Hansen, H., Almeida, A., Janssen, J. J. W. M., Mayer, J., Koskenvesa, P., Panayiotidis, P., Olsson-Strömberg, U., Martinez-Lopez, J., Rousselot, P., Vestergaard, H., Ehrencrona, H., Kairisto, V., Machová Poláková, K., Müller, M. C., Mustjoki, S., Berger, M. G., … EURO-SKI investigators (2018). Discontinuation of tyrosine kinase inhibitor therapy in chronic myeloid leukaemia (EURO-SKI): a prespecified interim analysis of a prospective, multicentre, non-randomised, trial. The Lancet. Oncology, 19(6), 747–757. https://doi.org/10.1016/S1470-2045(18)30192-X
- Eden RE, Coviello JM. Chronic Myelogenous Leukemia. [Updated 2023 Jan 16]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2026 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK531459/
- Khoury, J. D., Solary, E., Abla, O., Akkari, Y., Alaggio, R., Apperley, J. F., Bejar, R., Berti, E., Busque, L., Chan, J. K. C., Chen, W., Chen, X., Chng, W. J., Choi, J. K., Colmenero, I., Coupland, S. E., Cross, N. C. P., De Jong, D., Elghetany, M. T., Takahashi, E., … Hochhaus, A. (2022). The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia, 36(7), 1703–1719. https://doi.org/10.1038/s41375-022-01613-1
- Arber, D. A., Orazi, A., Hasserjian, R. P., Borowitz, M. J., Calvo, K. R., Kvasnicka, H. M., Wang, S. A., Bagg, A., Barbui, T., Branford, S., Bueso-Ramos, C. E., Cortes, J. E., Dal Cin, P., DiNardo, C. D., Dombret, H., Duncavage, E. J., Ebert, B. L., Estey, E. H., Facchetti, F., Foucar, K., … Tefferi, A. (2022). International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood, 140(11), 1200–1228. https://doi.org/10.1182/blood.2022015850
- Soverini, S., Bavaro, L., De Benedittis, C., Martelli, M., Iurlo, A., Orofino, N., Sica, S., Sorà, F., Lunghi, F., Ciceri, F., Galimberti, S., Baratè, C., Bonifacio, M., Scaffidi, L., Castagnetti, F., Gugliotta, G., Albano, F., Russo Rossi, A. V., Stagno, F., di Raimondo, F., … Martinelli, G. (2020). Prospective assessment of NGS-detectable mutations in CML patients with nonoptimal response: the NEXT-in-CML study. Blood, 135(8), 534–541. https://doi.org/10.1182/blood.2019002969
- Bochicchio, M. T., Petiti, J., Berchialla, P., Izzo, B., Giugliano, E., Ottaviani, E., Errichiello, S., Rege-Cambrin, G., Venturi, C., Luciano, L., Daraio, F., Calistri, D., Rosti, G., Saglio, G., Martinelli, G., Pane, F., Cilloni, D., Gottardi, E. M., & Fava, C. (2021). Droplet Digital PCR for BCR-ABL1 Monitoring in Diagnostic Routine: Ready to Start?. Cancers, 13(21), 5470. https://doi.org/10.3390/cancers13215470
- Pfirrmann, M., Baccarani, M., Saussele, S., Guilhot, J., Cervantes, F., Ossenkoppele, G., Hoffmann, V. S., Castagnetti, F., Hasford, J., Hehlmann, R., & Simonsson, B. (2016). Prognosis of long-term survival considering disease-specific death in patients with chronic myeloid leukemia. Leukemia, 30(1), 48–56. https://doi.org/10.1038/leu.2015.261
- Aghel, N., & Lipton, J. H. (2025). Cardiovascular Disease in Patients With Chronic Myeloid Leukemia: JACC: CardioOncology State-of-the-Art Review. JACC. CardioOncology, 7(6), 668–682. https://doi.org/10.1016/j.jaccao.2025.06.007