Key Takeaways
Primary myelofibrosis is a rare, chronic blood cancer in which the bone marrow becomes scarred (fibrosed), blood cell production goes haywire, and the spleen typically enlarges. It is one of three classic Philadelphia-negative myeloproliferative neoplasms (MPNs), alongside essential thrombocythemia and polycythemia vera.
- Pathogenesis ▾: Caused by clonal mutations in a hematopoietic stem cell, leading to overproduction of abnormal blood cells and bone marrow fibrosis due to inflammatory mediators.
- Laboratory investigations ▾: Complete blood count, peripheral blood smear, bone marrow aspiration and biopsy, molecular testing.
- Diagnostic criteria: Diagnosis follows the 2022 WHO and International Consensus Classification criteria, which require a bone marrow biopsy, exclusion of other myeloid cancers, and molecular testing.
- Treatment and Management ▾: Treatment is guided by risk score, not by the diagnosis alone. Low-risk patients are usually observed. Symptomatic or higher-risk patients receive JAK inhibitors, with allogeneic stem cell transplant as the only curative option.
*Click ▾ for more information
Introduction
Primary myelofibrosis is rare, but it shows up across hematology rotations and oncology wards because it teaches almost every key concept in the field at once: clonal stem cell biology, cytokine-driven inflammation, marrow failure, and risk-adapted therapy.
Three things define primary myelofibrosis:
- Abnormal blood cell production. The bone marrow makes too many of the wrong cells, in the wrong proportions.
- Bone marrow fibrosis. Scar tissue gradually fills the marrow, crowding out normal blood cell production.
- Splenomegaly. The spleen takes over some of the marrow's job and swells, sometimes dramatically.
Where it sits among blood cancers
Primary myelofibrosis is classified as a myeloproliferative neoplasm (MPN), a family of cancers in which the marrow overproduces one or more blood cell lines. The other classic MPNs are:
The "primary" part of the name matters. It tells you the disease arose on its own. When fibrosis develops on the back of ET or PV instead, it is called secondary (post-ET or post-PV) myelofibrosis.
Pathogenesis and Pathophysiology

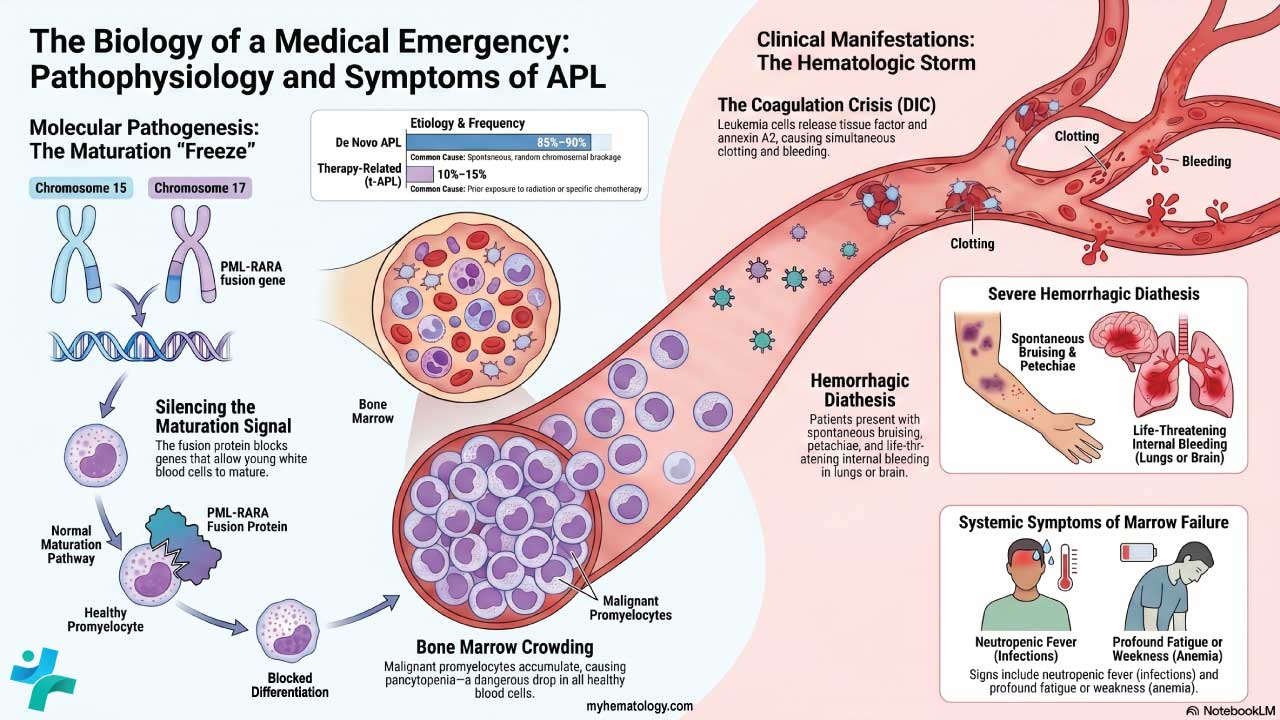
Primary myelofibrosis starts with an acquired mutation in a single hematopoietic stem cell — the master cell that gives rise to all blood cells. The mutation is not inherited. It happens at some point during life and gives the affected cell a growth advantage.
The mutated cell and its descendants overproduce blood cells, especially megakaryocytes. These abnormal megakaryocytes release inflammatory signals. The signals stimulate fibroblasts, the local connective-tissue cells, to lay down excessive collagen and reticulin fibers. That is the scar tissue you see on biopsy.
Three driver mutations dominate:
- JAK2 V617F mutation — found in roughly 50 to 60% of patients [1]. It locks the JAK2 protein in the "on" position, so the JAK-STAT signaling pathway runs constantly and pushes cells to keep dividing.
- CALR (calreticulin) mutation — present in about 25 to 35%. CALR Type 1 mutations carry a relatively favorable prognosis [1]. Recent developments have shifted CALR from being just a prognostic marker to an actionable therapeutic target. Investigational therapies like INCA033989, a novel monoclonal antibody directly targeting mutant CALR on hematopoietic stem cells, have shown promising potential to prevent downstream JAK-STAT activation and reduce the mutant allele burden [10,11].
- MPL mutation — found in about 5 to 10%. MPL codes for the thrombopoietin receptor, and mutations leave it overactive.
Around 10 to 15% of patients are triple-negative for these three drivers. They need broader genetic profiling to look for mutations such as ASXL1 or EZH2, and they tend to have a worse prognosis [1,2].
What sparks these mutations in the first place is largely unknown. Long-term exposure to benzene or high doses of ionizing radiation has been linked to higher risk, but most patients have no identifiable trigger.
Epidemiology
Primary myelofibrosis is uncommon. Key figures to know:
- Prevalence: roughly 1 to 2 cases per 100,000 people [6].
- Median age at diagnosis: about 65 to 70 years.
- Sex distribution: slightly more common in men, with a male-to-female ratio of about 1.5 to 1.
- Geography: rates are modestly higher in Europe and North America than in other regions.
Risk Factors
The strongest risk factor is age. The disease is rare before 50 and rises steadily after that.
Other contributors are:
- Chemical exposure, particularly long-term contact with benzene and certain industrial solvents.
- Ionizing radiation, especially at high doses (occupational, therapeutic, or historical events such as atomic-bomb survivors).
- Family history of any MPN. The absolute risk is still low, but it is higher than in the general population.
Signs and Symptoms of PMF
Early primary myelofibrosis is often silent. Many patients are picked up incidentally on a routine blood test that shows mild anemia or an unexpectedly large spleen. As the disease progresses, three symptom clusters tend to dominate.
Constitutional symptoms
These reflect the inflammatory cytokine surge that comes with the disease:
- Persistent fatigue and weakness.
- Drenching night sweats.
- Low-grade fever.
- Unintended weight loss.
Symptoms of marrow failure
As fibrosis crowds out healthy marrow, blood cell counts fall:
- Anemia causes pallor, breathlessness, and dizziness.
- Thrombocytopenia (low platelets) leads to easy bruising, nosebleeds, and gum bleeding.
- Neutropenia raises the risk of infection.
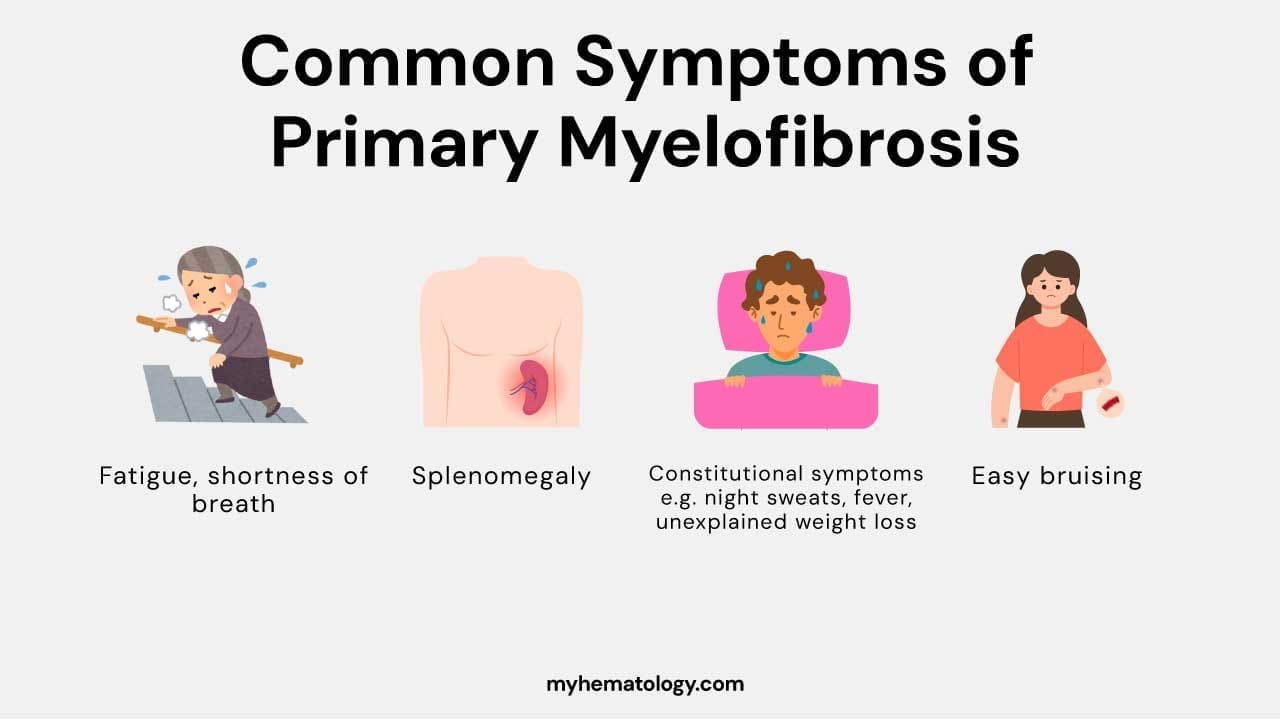
This image illustrates several key signs and symptoms associated with primary myelofibrosis (PMF). Anemia, characterized by a decrease in red blood cells, can lead to fatigue, weakness, and shortness of breath. Massive hepatosplenomegaly refers to a significant enlargement of both the liver (hepatomegaly) and spleen (splenomegaly), which can cause discomfort, early satiety (feeling full after eating a small amount), and other complications. Additionally, hypermetabolism (increased metabolic rate) can manifest as unintended weight loss, night sweats, and fatigue, potentially contributing to a reduced quality of life in individuals with PMF.
Symptoms of splenomegaly
The spleen takes over some blood cell production through extramedullary hematopoiesis. As it enlarges, it presses on neighboring organs:
- A dragging or full sensation in the upper-left abdomen.
- Early satiety (feeling full after only a few bites).
- Bone pain, often in the lower back, ribs, or legs.
Complications
Beyond the symptoms above, primary myelofibrosis can produce several serious complications:
- Infection. Low or dysfunctional white cells weaken immunity. Bacterial, viral, and fungal infections are all more likely.
- Transformation to acute myeloid leukemia (AML). About 10 to 20% of patients eventually progress to AML (also called blast-phase MPN). Median survival after transformation is only around 5 to 6 months unless the patient can bridge to a stem cell transplant [1].
- Bleeding and thrombosis. Patients can have either, sometimes both. Deep vein thrombosis and pulmonary embolism are particularly important to recognize.
- Portal hypertension. A massive spleen can choke blood flow through the portal vein, leading to ascites and bleeding from esophageal or gastric varices.
- Iron overload. Repeated red cell transfusions deposit iron in the heart, liver, and endocrine glands. Iron chelation is usually considered after roughly 20 transfusions or when ferritin exceeds 1000 ng/mL.
Laboratory Investigations
A diagnostic workup for primary myelofibrosis layers three sources of information: peripheral blood, bone marrow histology, and molecular genetics. Each step narrows the differential.
Complete blood count (CBC)
CBC findings shift as the disease evolves:
- Anemia, usually normocytic and normochromic, is the most common cytopenia and worsens as fibrosis progresses.
- White cells can be high in early disease (leukocytosis) and low later, when the marrow fails.
- Platelets often run high early (thrombocytosis) and crash to severe thrombocytopenia in advanced disease.
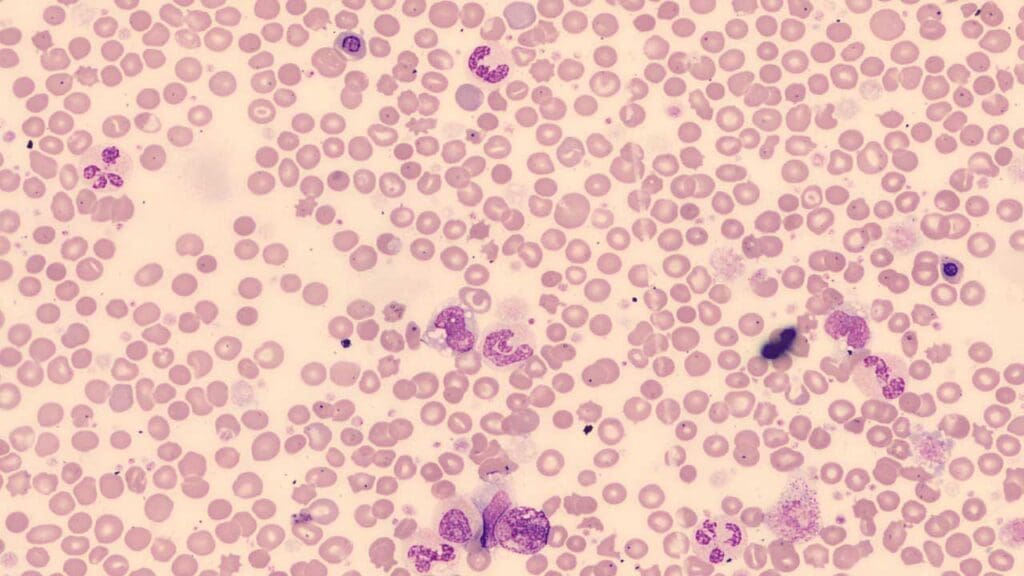
Peripheral blood smear
A peripheral blood smear is a cheap, fast tool that can flag PMF. The classic triad to look for is:
- Leukoerythroblastosis — immature white cells (myelocytes, metamyelocytes) and nucleated red cells appearing in the circulation. This signals that fibrosis has disrupted the normal marrow-blood barrier.
- Dacrocytes (teardrop cells) — red cells distorted into a teardrop shape as they squeeze through scarred marrow or an enlarged spleen.
- Atypical platelets — giant, hypogranular forms and circulating megakaryoblast fragments.
Bone marrow aspiration and biopsy
Bone marrow evaluation is the gold standard for diagnosing primary myelofibrosis [1,6].
- Aspiration often produces a dry tap: no liquid marrow can be drawn because reticulin and collagen have packed the marrow space.
- Biopsy morphology shows clustered, atypical megakaryocytes with hyperlobulated nuclei. Cellularity is high in early disease and drops later.
- Reticulin staining is mandatory and grades fibrosis from MF-0 to MF-3.

WHO/European Consensus Fibrosis Grading
| Grade | Fiber pattern | Clinical interpretation |
|---|---|---|
| MF-0 | Scattered linear reticulin, no intersections | Normal marrow |
| MF-1 | Loose reticulin network with many intersections | Prefibrotic PMF |
| MF-2 | Diffuse, dense reticulin with extensive intersections; occasional collagen or osteosclerosis | Overt PMF |
| MF-3 | Dense reticulin with coarse collagen bundles; significant osteosclerosis | Advanced overt PMF |
Molecular testing
Molecular testing looks for the three driver mutations (JAK2, CALR, MPL) and, where available, additional high-risk mutations such as ASXL1, EZH2, SRSF2, U2AF1, and IDH1/2. These results feed directly into modern risk scoring [1].

Diagnostic Criteria
The 2022 WHO and International Consensus Classification (ICC) split the disease into two phases: prefibrotic PMF (pre-PMF) and overt PMF. Diagnosis requires all three major criteria plus at least one minor criterion [2].
Overt PMF — major criteria (all three required)
- Bone marrow morphology: megakaryocytic proliferation and atypia, with reticulin and/or collagen fibrosis grade 2 or 3.
- Exclusion of other myeloid neoplasms: does not meet criteria for ET, PV, BCR-ABL1–positive CML, MDS, or another myeloid cancer.
- Molecular evidence: a JAK2, CALR, or MPL mutation. In triple-negative cases, another clonal marker (such as ASXL1 or EZH2) or absence of reactive marrow fibrosis fulfills this criterion.
Overt PMF — minor criteria (at least one, confirmed on two occasions)
- Anemia not explained by another condition.
- WBC count ≥ 11 × 10⁹/L.
- Palpable splenomegaly.
- LDH above the institution's upper limit of normal.
- Leukoerythroblastosis on the peripheral smear.
Prefibrotic PMF (pre-PMF)
The criteria mirror overt PMF with one key difference: the marrow shows megakaryocyte atypia without fibrosis above MF-1. Cellularity is usually high, with prominent granulopoiesis. Leukoerythroblastosis is not required as a minor criterion at this stage.
Treatment and Management
Primary myelofibrosis treatment is guided by risk score, not by the diagnosis alone. This is the most important principle to take away. Two patients with the same biopsy findings can need very different management.
Risk stratification
Before starting any treatment, patients are scored using DIPSS-Plus (clinical) or the more modern, molecularly informed MIPSS70+ v2.0 [1]. The score sets the goal:
- Low risk: maximize quality of life and watch for progression.
- High risk: extend survival and evaluate for stem cell transplantation.
Asymptomatic and low-risk disease: watch and wait
For low-risk or intermediate-1 patients without bothersome symptoms, severe anemia, or massive splenomegaly, active observation is the standard of care. Starting JAK inhibitors early in this group exposes patients to side effects without proven survival benefit.
JAK inhibitors
For symptomatic patients, especially those with constitutional symptoms or a painful enlarged spleen, JAK inhibitors are the cornerstone of medical treatment. They do not cure the disease or fully reverse fibrosis, but they can shrink the spleen and dampen inflammation.
Managing these agents in the real world is highly dynamic. For example, the vast majority of patients on ruxolitinib require proactive dose adjustments or interruptions during their treatment course due to fluctuating blood counts or emerging cytopenias [4].
Choice of agent depends on the dominant clinical problem:
- Ruxolitinib (Jakafi) — JAK1/JAK2 inhibitor. The first-line standard for intermediate or high-risk primary myelofibrosis with symptomatic splenomegaly. It can worsen anemia and thrombocytopenia [4].
- Fedratinib (Inrebic) — selective JAK2 inhibitor. Used in patients intolerant of, or relapsed after, ruxolitinib. Carries a black box warning for Wernicke's encephalopathy; thiamine levels must be monitored.
- Pacritinib (Vonjo) — JAK2 and IRAK1 inhibitor. Specifically approved for patients with severe thrombocytopenia (platelets < 50 × 10⁹/L), where ruxolitinib is hard to dose [5].
- Momelotinib (Ojjaara) — JAK1, JAK2, and ACVR1 inhibitor. FDA-approved in September 2023 for intermediate or high-risk myelofibrosis with anemia. By inhibiting ACVR1, it lowers hepcidin, restores iron handling, and improves red cell production [7,8].
Managing cytopenias
When marrow failure dominates the clinical picture, treatment shifts toward supporting individual cell lines.
Anemia
- Erythropoiesis-stimulating agents (epoetin alfa, darbepoetin), useful when endogenous erythropoietin is below 500 mU/mL.
- Danazol, a synthetic androgen that can boost red cell production in some patients.
- Luspatercept, an erythroid maturation agent increasingly used for transfusion-dependent anemia in MPNs.
- Red cell transfusions for severe symptomatic anemia. Iron chelation therapy is added to manage the long-term iron overload that follows.
High white cells or platelets
- Hydroxyurea, a cytoreductive agent that controls leukocytosis or thrombocytosis and modestly shrinks the spleen.
Refractory splenomegaly
If targeted therapy and cytoreduction fail to control a massive, painful spleen, mechanical interventions are considered:
- Splenectomy. High risk in primary myelofibrosis because of bleeding, thrombosis, and major compensatory liver enlargement. Reserved for last-line situations.
- Splenic irradiation. Low-dose radiation can give 3 to 6 months of relief but often causes prolonged, severe cytopenias.
Allogeneic stem cell transplantation
Allogeneic stem cell transplantation (allo-HSCT) is the only treatment that can cure primary myelofibrosis [1]. It replaces the diseased marrow with healthy donor stem cells. Because the procedure carries a real risk of death from graft-versus-host disease and infection, patient selection is strict:
- Intermediate-2 or high-risk disease.
- Younger, fitter patients (typically under 70 to 75) without major comorbidities.
- A suitable donor.
JAK inhibitors are often used as a bridge to transplant, shrinking the spleen and improving the patient's condition before the procedure. Current guidance favors early transplant evaluation in eligible high-risk patients, before comorbidities accumulate.
Recent prospective clinical trial data has strongly reinforced this approach. The multicenter RuxoAllo study compared patients who underwent stem cell transplantation after three months of ruxolitinib induction against those who continued ruxolitinib indefinitely due to a lack of a donor. The transplant cohort demonstrated significantly improved event-free survival (EFS), validating that early transplant evaluation is the optimal path for eligible intermediate-2 or high-risk patients before comorbidities multiply [15].
Emerging combination therapies
The current frontier in primary myelofibrosis treatment is combination therapy, particularly adding a second agent to ruxolitinib in JAK-inhibitor-naive patients.
- BET inhibitors (pelabresib). The phase 3 MANIFEST-2 trial has demonstrated that combining the BET inhibitor pelabresib with ruxolitinib provides deeper and more durable disease control for JAK-inhibitor-naive patients. Updated 96-week data showed that 91.5% of evaluable patients on the combination therapy maintained at least a 35% reduction in spleen volume, compared to just 57.6% on ruxolitinib alone. The combination also yielded significantly better sustained symptom improvement [12].
- TGF-β superfamily inhibitors (elritercept). For patients suffering from severe cytopenias (anemia and thrombocytopenia), a novel agent called elritercept is currently under advanced clinical investigation. Elritercept is an activin inhibitor designed to block a subset of the transforming growth factor beta (TGF-β) family of proteins, promoting hematopoiesis to restore natural red blood cell and platelet production either as a single agent or alongside ruxolitinib [13,14].
- BCL-xL inhibitors (navitoclax). Target apoptotic pathways to push mutated clones to die.
Symptom assessment in practice
Most modern trials and clinics use the MPN-SAF Total Symptom Score (TSS) to track how a patient is feeling over time. It is a 10-item self-report covering fatigue, abdominal discomfort, early satiety, inactivity, concentration problems, night sweats, itching, bone pain, fever, and weight loss. A drop in TSS is one of the standard endpoints used to judge treatment response.
Prognosis
Modern prognostic scoring depends partly on whether the patient is a transplant candidate.
MIPSS70+ v2.0 (the integrated standard)
This is the most-used model for transplant-eligible patients (usually under 70). It weighs molecular data alongside clinical severity [1].
Prognostic Scoring
| Risk category | Total points | Median overall survival | 10-year OS |
|---|---|---|---|
| Very low | 0 | Not reached | ~92% |
| Low | 1–2 | ~16.4 years | ~56% |
| Intermediate | 3–4 | ~7.7 years | ~37% |
| High | 5–8 | ~4.1 years | ~13% |
| Very high | ≥9 | ~1.8 years | <5% |
GIPSS (Genetically Inspired Prognostic Scoring System)
GIPSS uses only mutations and karyotype, which makes it useful when blood counts and symptoms are fluctuating [1]:
- Low risk: median survival of ~26 years.
- High risk: median survival of ~2.6 years.
DIPSS-Plus
Still widely used at the bedside. It tallies eight clinical risk factors: age >65, Hb <10 g/dL, WBC >25 × 10⁹/L, blasts ≥1%, constitutional symptoms, unfavorable karyotype, platelets <100 × 10⁹/L, and red cell transfusion dependency.
High molecular risk (HMR) mutations
Certain mutations push patients into a higher risk tier no matter what their blood counts say. The presence of two or more HMR mutations alone shortens survival significantly [1]:
- ASXL1 — the most common HMR mutation; strongly linked to shorter survival and fibrotic progression.
- SRSF2 and U2AF1 (Q157) — spliceosome mutations that raise the risk of leukemic transformation.
- EZH2 — linked to aggressive disease and poor outcomes.
- IDH1/IDH2 — associated with rapid progression to blast phase.
CALR Type 1 mutations are the bright spot. They generally predict longer survival and lower thrombosis risk.
Risk of leukemic transformation
About 10 to 20% of primary myelofibrosis patients progress to secondary AML, also called PMF in blast phase [1]. Transformation often happens within the first 5 years if HMR mutations such as SRSF2 or IDH1/2 are present. Once in blast phase, median survival is roughly 5 to 6 months unless the patient can move quickly to a stem cell transplant.
Summary of Prognosis
| Factor | Favorable Prognosis | Poor Prognosis |
|---|---|---|
|
Genetics
|
||
|
Karyotype
|
||
|
Clinical
|
||
|
Spleen
|
Post-ET and Post-PV Myelofibrosis
Some patients first diagnosed with another MPN go on to develop fibrosis years later.
- Post-ET myelofibrosis occurs when essential thrombocythemia progresses to a fibrotic phase. The transformation usually takes 7 to 20 years from the initial ET diagnosis. Lifetime risk is about 1.6 to 9%.
- Post-PV myelofibrosis occurs when polycythemia vera evolves the same way. Lifetime risk is somewhat higher, in the 5 to 14% range.
Treatment principles overlap with primary myelofibrosis, but the prior disease history shapes monitoring, transplant timing, and prognosis.
Frequently Asked Questions (FAQs)
Is primary myelofibrosis curable?
Allogeneic stem cell transplantation is the only treatment that can cure primary myelofibrosis. It is reserved for younger, fitter patients with intermediate-2 or high-risk disease, because the procedure itself carries a meaningful risk of death from infection or graft-versus-host disease. All other treatments, including JAK inhibitors, aim to control symptoms and improve quality of life rather than cure the disease.
What is the difference between primary and secondary myelofibrosis?
Primary myelofibrosis develops on its own from a stem cell mutation in the bone marrow. Secondary myelofibrosis evolves from an existing myeloproliferative neoplasm, most often essential thrombocythemia (post-ET MF) or polycythemia vera (post-PV MF). The transition usually takes 7 to 20 years. Treatment principles are similar, but the prior disease history shapes prognosis and monitoring.
How is primary myelofibrosis diagnosed?
Diagnosis follows the 2022 WHO and International Consensus Classification criteria. Doctors look for three major findings: characteristic megakaryocyte changes with bone marrow fibrosis, exclusion of other myeloid cancers, and a driver mutation in JAK2, CALR, or MPL. At least one minor criterion must also be present, such as anemia, an enlarged spleen, raised LDH, leukocytosis, or leukoerythroblastosis on a blood smear.
Which JAK inhibitor is best for myelofibrosis?
The choice depends on the dominant problem. Ruxolitinib is first-line for most patients with adequate platelet counts. Fedratinib is used after ruxolitinib fails or is not tolerated. Pacritinib is preferred when platelets are very low (under 50 × 10⁹/L). Momelotinib is the go-to when anemia dominates the picture, because it also lowers hepcidin and improves red cell production.
What does a "triple-negative" diagnosis mean?
Triple-negative means the patient does not carry any of the three classic driver mutations (JAK2, CALR, or MPL). About 10 to 15% of primary myelofibrosis patients fall into this group. These cases need broader genetic testing to look for other mutations such as ASXL1 or EZH2, and they generally carry a more guarded prognosis than CALR-mutated disease.
How does primary myelofibrosis affect survival?
Survival depends heavily on risk stratification. Using the MIPSS70+ v2.0 system, very-low-risk patients have a 10-year survival above 90%, while very-high-risk patients have a median survival under two years. About 10 to 20% of patients eventually transform to acute myeloid leukemia, which carries a much shorter prognosis unless followed quickly by a stem cell transplant.
Glossary of Related Medical Terms
- Allogeneic stem cell transplantation (allo-HSCT): A procedure that replaces diseased bone marrow with healthy donor stem cells. Currently the only treatment that can cure primary myelofibrosis.
- Constitutional symptoms: Whole-body symptoms caused by inflammation, including fatigue, drenching night sweats, fever, and unintended weight loss.
- Cytopenia: A low count of one or more types of blood cell.
- Dacrocyte (teardrop cell): A red blood cell shaped like a teardrop, common in conditions where the bone marrow is fibrotic.
- DIPSS-Plus: A clinical tool that estimates survival in primary myelofibrosis using age, blood counts, symptoms, and other markers.
- Driver mutation: A genetic change that directly causes a cancer to develop. In primary myelofibrosis the driver mutations are JAK2, CALR, and MPL.
- Erythropoiesis: The body's production of red blood cells.
- Extramedullary hematopoiesis: Blood cell production happening outside the bone marrow, usually in the spleen or liver.
- Fibroblast: A connective tissue cell that produces collagen. Overactive fibroblasts in the bone marrow drive primary myelofibrosis scarring.
- Hematopoietic stem cell: The master cell in the bone marrow that gives rise to all blood cells.
- Hepcidin: A hormone that controls how the body uses iron. High levels worsen anemia by trapping iron away from red blood cell production.
- JAK-STAT pathway: A cell signaling system that controls growth and division. Overactive signaling drives the abnormal blood cell production seen in primary myelofibrosis.
- Leukoerythroblastosis: A blood smear pattern showing immature white cells and nucleated red cells in the circulation.
- Megakaryocyte: A large bone marrow cell that produces platelets. Atypical, clustered megakaryocytes are a defining feature of primary myelofibrosis.
- MIPSS70+ v2.0: An updated risk tool that combines clinical features with molecular data.
- Myeloproliferative neoplasm (MPN): A group of blood cancers in which the bone marrow makes too many of one or more types of blood cell.
- Osteosclerosis: Abnormal hardening and thickening of bone, sometimes seen alongside marrow fibrosis on biopsy.
- Reticulin: A type of fine, collagen-like fiber. The amount of reticulin in the marrow is graded MF-0 to MF-3.
- Splenomegaly: Enlargement of the spleen, the most visible physical sign of primary myelofibrosis.
- Thrombocytopenia: A low platelet count, which raises the risk of bleeding.
- Triple-negative PMF: Primary myelofibrosis without any of the three driver mutations (JAK2, CALR, MPL).
Disclaimer: This article is intended for educational and informational purposes only. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Tefferi A. (2023). Primary myelofibrosis: 2023 update on diagnosis, risk-stratification, and management. American journal of hematology, 98(5), 801–821. https://doi.org/10.1002/ajh.26857
- Gianelli, U., Thiele, J., Orazi, A., Gangat, N., Vannucchi, A. M., Tefferi, A., & Kvasnicka, H. M. (2023). International Consensus Classification of myeloid and lymphoid neoplasms: myeloproliferative neoplasms. Virchows Archiv : an international journal of pathology, 482(1), 53–68. https://doi.org/10.1007/s00428-022-03480-8
- Rampal, R. K., Grosicki, S., Chraniuk, D., Abruzzese, E., Bose, P., Gerds, A. T., Vannucchi, A. M., Palandri, F., Lee, S. E., Gupta, V., Lucchesi, A., Oh, S. T., Kuykendall, A. T., Patriarca, A., Álvarez-Larrán, A., Mesa, R., Kiladjian, J. J., Talpaz, M., Scandura, J. M., Lavie, D., … Mascarenhas, J. (2025). Pelabresib plus ruxolitinib for JAK inhibitor-naive myelofibrosis: a randomized phase 3 trial. Nature medicine, 31(5), 1531–1538. https://doi.org/10.1038/s41591-025-03572-3
- Verstovsek, S., Mesa, R. A., Livingston, R. A., Hu, W., & Mascarenhas, J. (2023). Ten years of treatment with ruxolitinib for myelofibrosis: a review of safety. Journal of hematology & oncology, 16(1), 82. https://doi.org/10.1186/s13045-023-01471-z
- Mascarenhas, J., Hoffman, R., Talpaz, M., Gerds, A. T., Stein, B., Gupta, V., Szoke, A., Drummond, M., Pristupa, A., Granston, T., Daly, R., Al-Fayoumi, S., Callahan, J. A., Singer, J. W., Gotlib, J., Jamieson, C., Harrison, C., Mesa, R., & Verstovsek, S. (2018). Pacritinib vs Best Available Therapy, Including Ruxolitinib, in Patients With Myelofibrosis: A Randomized Clinical Trial. JAMA oncology, 4(5), 652–659. https://doi.org/10.1001/jamaoncol.2017.5818
- Takenaka, K., Shimoda, K., & Akashi, K. (2018). Recent advances in the diagnosis and management of primary myelofibrosis. The Korean journal of internal medicine, 33(4), 679–690. https://doi.org/10.3904/kjim.2018.033
- Mesa, R. A., Kiladjian, J. J., Catalano, J. V., Devos, T., Egyed, M., Hellmann, A., McLornan, D., Shimoda, K., Winton, E. F., Deng, W., Dubowy, R. L., Maltzman, J. D., Cervantes, F., & Gotlib, J. (2017). SIMPLIFY-1: A Phase III Randomized Trial of Momelotinib Versus Ruxolitinib in Janus Kinase Inhibitor-Naïve Patients With Myelofibrosis. Journal of clinical oncology : official journal of the American Society of Clinical Oncology, 35(34), 3844–3850. https://doi.org/10.1200/JCO.2017.73.4418
- Verstovsek, S., Gerds, A. T., Vannucchi, A. M., Al-Ali, H. K., Lavie, D., Kuykendall, A. T., Grosicki, S., Iurlo, A., Goh, Y. T., Lazaroiu, M. C., Egyed, M., Fox, M. L., McLornan, D., Perkins, A., Yoon, S. S., Gupta, V., Kiladjian, J. J., Granacher, N., Lee, S. E., Ocroteala, L., … MOMENTUM Study Investigators (2023). Momelotinib versus danazol in symptomatic patients with anaemia and myelofibrosis (MOMENTUM): results from an international, double-blind, randomised, controlled, phase 3 study. Lancet (London, England), 401(10373), 269–280. https://doi.org/10.1016/S0140-6736(22)02036-0
- Oon, S. F., Singh, D., Tan, T. H., Lee, A., Noe, G., Burbury, K., & Paiva, J. (2019). Primary myelofibrosis: spectrum of imaging features and disease-related complications. Insights into imaging, 10(1), 71. https://doi.org/10.1186/s13244-019-0758-y
- Incyte Corporation. (2022). Incyte's Novel Mutant CALR Antibody Unveiled at ASH 2022 Plenary Scientific Session [Press release]. Retrieved from https://investor.incyte.com/news-releases/news-release-details/incytes-novel-mutant-calr-antibody-unveiled-ash-2022-plenary
- National Cancer Institute. (n.d.). Definition of anti-mutant calreticulin monoclonal antibody INCA033989. NCI Drug Dictionary. Retrieved from https://www.cancer.gov/publications/dictionaries/cancer-drug/def/anti-mutant-calreticulin-monoclonal-antibody-inca033989
- Oncodaily. (2025). MANIFEST-2 Trial: Pelabresib + Ruxolitinib in MF. Retrieved from https://oncodaily.com/oncolibrary/manifest-2-trial-pelabresib-ruxolitinib
- Takeda Oncology. (2024). Takeda Strengthens Oncology Pipeline with Elritercept through Licensing Agreement with Keros Therapeutics [Press release]. Retrieved from https://www.takedaoncology.com/newsroom/news-releases/2024/elritercept-licensing/
- U.S. National Library of Medicine. (n.d.). A Study of Elritercept Alone or Together With Ruxolitinib in Adults With Myelofibrosis. ClinicalTrials.gov. Identifier: NCT05037760. Retrieved from https://clinicaltrials.gov/study/NCT05037760
- Kröger, N., et al. (2025). Ruxolitinib versus allogeneic stem cell transplantation for patients with myelofibrosis according to donor availability: A prospective multicenter phase II trial. Blood (2025) 146 (Supplement 1): 911. https://doi.org/10.1182/blood-2025-911