TL;DR
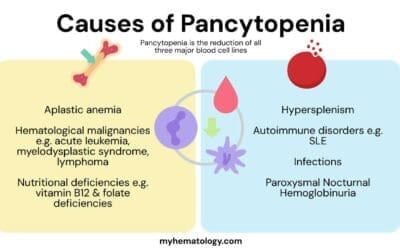
Bone marrow failure is the inability of the bone marrow to produce enough mature blood cells, leading to low counts (pancytopenia).
Causes ▾: The causes are broadly divided into acquired and inherited.
- Acquired causes include immune-mediated attacks (aplastic anemia), infections (like Parvovirus B19), drugs and toxins, radiation exposure, and nutritional deficiencies (B12, folate).
- Inherited causes are rare genetic disorders, such as Fanconi Anemia (DNA repair defects) and Dyskeratosis Congenita (telomere disorders).
- Anemia: leads to fatigue, shortness of breath, and pallor.
- Neutropenia: causes frequent and severe infections.
- Thrombocytopenia: results in easy bruising, tiny red spots on the skin (petechiae), and abnormal bleeding.
- Complete Blood Count (CBC) to show low cell counts and a reticulocyte count to confirm bone marrow underproduction.
- Bone Marrow Biopsy is the gold standard for diagnosis.
- Cytogenetic and molecular studies are essential to identify specific gene mutations, especially for MDS and inherited disorders.
Treatment & Management ▾: Treatment depends on the cause and severity.
- Supportive care is crucial and includes blood transfusions for anemia and thrombocytopenia, and growth factor support to raise white blood cell counts.
- Definitive treatments can include Immunosuppressive Therapy (IST) for aplastic anemia or Hematopoietic Stem Cell Transplantation (HSCT), which is the only curative option for many severe cases.
*Click ▾ for more information
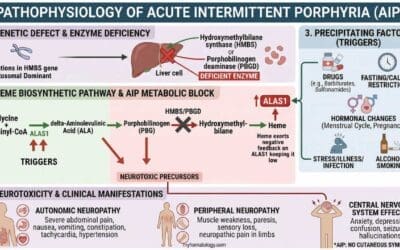
Bone marrow failure (BMF) represents a diverse group of hematologic conditions characterized by a deficiency in the production of one or more types of blood cells – red blood cells, white blood cells, and platelets. This pancytopenia can arise from a range of acquired or inherited disorders, all of which disrupt the normal function of hematopoietic stem cells (HSCs) within the bone marrow.
The Role of Hematopoietic Stem Cells in Health and Disease
The bone marrow is the body’s primary factory for blood cell production. Residing within this spongy tissue are HSCs, multipotent progenitor cells with the unique ability to self-renew and differentiate into all mature blood cell lineages.
A healthy bone marrow microenvironment, or niche, is essential for supporting these stem cells. It is composed of a complex network of stromal cells, growth factors, and extracellular matrix components. When this delicate balance is disturbed, either by an intrinsic defect in the HSCs themselves or by an extrinsic attack on the bone marrow microenvironment, bone marrow failure (BMF) can occur.
Classification of Bone Marrow Failure
Aplastic Anemia
This is the classic example of bone marrow failure. It is characterized by a “hypoplastic” or “empty” bone marrow, meaning the hematopoietic stem cells have been lost or suppressed. The result is a deficiency in the production of all three major blood cell types (red cells, white cells, and platelets), a condition known as pancytopenia. Most cases are idiopathic, meaning the cause is unknown, but they are often immune-mediated.
Myelodysplastic Syndromes (MDS)
In contrast to aplastic anemia, MDS is characterized by “ineffective hematopoiesis.”
Pathophysiology: MDS is a clonal hematopoietic stem cell disorder. Unlike aplastic anemia, the bone marrow in MDS is often hypercellular, but the cells are abnormal (dysplastic) and undergo programmed cell death (apoptosis) before they can fully mature and exit the marrow. This leads to “ineffective hematopoiesis,” where the bone marrow is producing cells, but they are defective and do not function properly. The underlying cause is genetic instability, leading to mutations in various genes involved in cell signaling and transcription, with common mutations in genes like SF3B1, TET2, and ASXL1. MDS is considered a pre-malignant condition due to its high risk of progression to acute myeloid leukemia (AML).
Clinical Guidelines: The diagnosis of MDS requires a detailed evaluation of peripheral blood and bone marrow morphology to identify dysplasia in one or more cell lines. Cytogenetic and molecular studies are essential to identify specific chromosomal abnormalities and gene mutations, as these are critical for both diagnosis and prognostication. The Revised International Prognostic Scoring System (IPSS-R) is a widely used tool that incorporates blast percentage, cytogenetics, and the number and severity of cytopenias to stratify patients into risk groups, which then guides treatment decisions. Treatment ranges from supportive care with transfusions for low-risk patients to hypomethylating agents (e.g., azacitidine, decitabine) for higher-risk individuals, and in some cases, allogeneic HSCT.
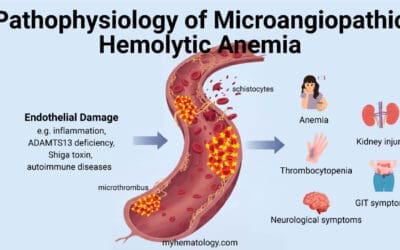
Pancytopenia
This is a clinical finding, not a specific disease, defined as a reduction in the number of red blood cells, white blood cells, and platelets. While it is a key feature of aplastic anemia and can be present in advanced MDS, it can also be caused by other conditions that infiltrate or suppress the bone marrow, such as leukemia, lymphoma, or certain infections. Therefore, it is important to distinguish the underlying cause of pancytopenia.
Causes of Bone Marrow Failure
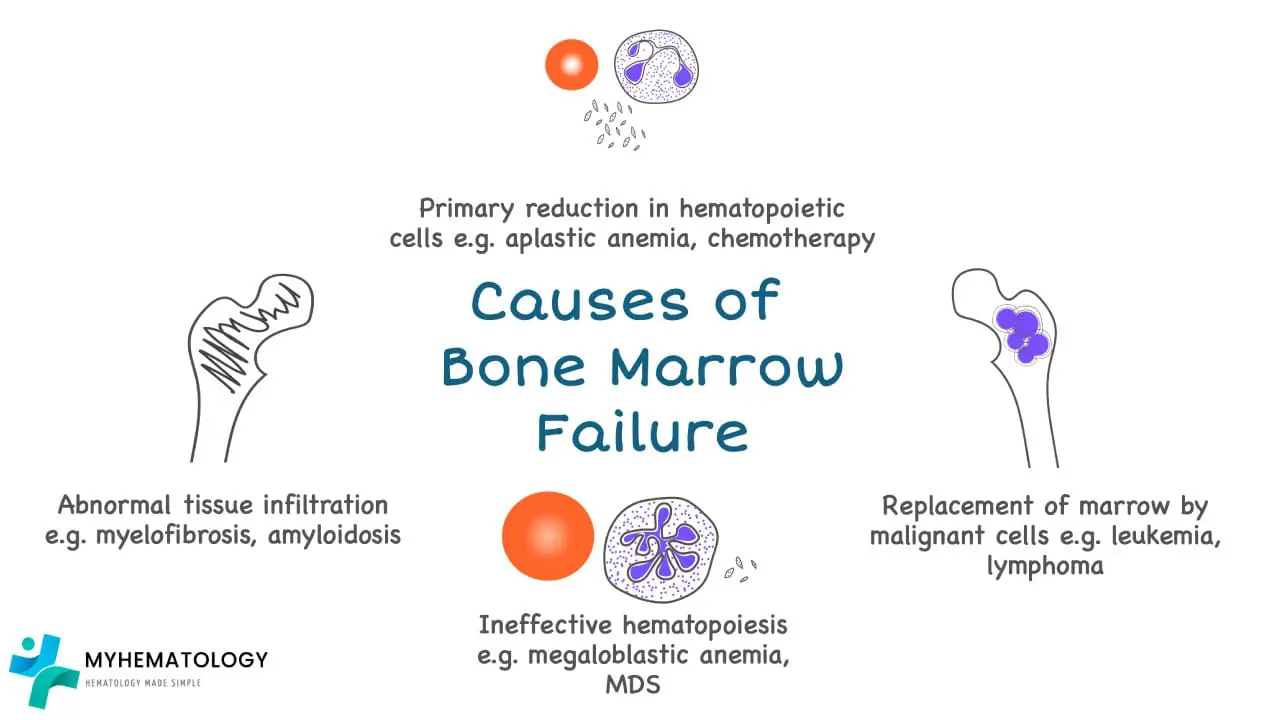
Acquired Causes of Bone Marrow Failure
The majority of bone marrow failure (BMF) cases are acquired, meaning they are not inherited from a parent. They often result from an immune-mediated attack, exposure to toxins, or the development of clonal disorders.
Acquired Aplastic Anemia (Immune-mediated)
Pathophysiology: AAA is the classic example of immune-mediated bone marrow failure (BMF). It is a rare and severe condition in which a patient’s own cytotoxic T lymphocytes (CD8+ T cells) mistakenly attack and destroy the HSCs in the bone marrow. This autoimmune process is often triggered by an unknown viral infection, drug exposure, or an environmental toxin in a genetically susceptible individual. The result is a profoundly hypocellular bone marrow unable to produce an adequate number of blood cells.
Clinical Guidelines: Diagnosis relies on bone marrow biopsy showing marked hypocellularity (typically <25%) with an absence of abnormal cells or fibrosis. Treatment is guided by disease severity. For young, severely ill patients, a hematopoietic stem cell transplant (HSCT) from a matched sibling donor is the gold standard. For those without a suitable donor, or who are not transplant candidates, immunosuppressive therapy (IST) with anti-thymocyte globulin (ATG) and cyclosporine is the primary treatment.
Infections
Pathophysiology: Certain viruses, most notably Parvovirus B19, directly infect and destroy erythroid progenitor cells, leading to severe aplastic crisis in patients with underlying hemolytic disorders. Other viruses like HIV, Epstein-Barr virus (EBV), and Hepatitis viruses can cause bone marrow suppression through direct cytopathic effects, immune-mediated mechanisms, or by altering the bone marrow microenvironment.
Clinical Guidelines: A comprehensive infectious disease workup, including viral serology and polymerase chain reaction (PCR) tests, is crucial to identify these triggers and guide specific antiviral therapy if available.
Drugs and Toxins
Pathophysiology: These can cause bone marrow failure through two main mechanisms. Dose-dependent toxicity, as seen with chemotherapeutic agents like alkylating agents or antimetabolites, directly damages the DNA of rapidly dividing hematopoietic stem cells. Idiosyncratic toxicity is an unpredictable, non-dose-dependent reaction that occurs in a small number of susceptible individuals, such as with the antibiotic chloramphenicol or the NSAID phenylbutazone. Environmental toxins like benzene and pesticides can also cause similar damage.
Clinical Guidelines: A detailed history of all medications and environmental exposures is critical. Discontinuation of the suspected agent is the first and most important step in management.
Radiation Exposure
Pathophysiology: Ionizing radiation causes extensive DNA damage to hematopoietic stem cells, leading to cell death (apoptosis) and an inability to proliferate. The degree of bone marrow failure is directly related to the dose of radiation received and the area of the body exposed.
Clinical Guidelines: Management involves immediate supportive care, including blood transfusions and granulocyte colony-stimulating factor (G-CSF) to stimulate any remaining functional stem cells.
Nutritional Deficiencies
Pathophysiology: Severe deficiencies of Vitamin B12 and folate impair DNA synthesis, which is essential for the rapid proliferation of hematopoietic cells. This leads to ineffective hematopoiesis, where cells grow but cannot divide properly, resulting in large, immature red blood cells (megaloblastic anemia) and pancytopenia.
Clinical Guidelines: Diagnosis is confirmed by measuring serum Vitamin B12 and folate levels. Treatment involves prompt and appropriate supplementation, which can rapidly reverse the hematologic abnormalities.
Myelofibrosis and other Infiltrative Disorders
Pathophysiology: Myelofibrosis is a type of myeloproliferative neoplasm (MPN) characterized by the progressive replacement of normal hematopoietic tissue with fibrous, scar-like connective tissue. This process is driven by the excessive release of fibrogenic cytokines from abnormal megakaryocytes. The dense scar tissue physically “crowds out” hematopoietic stem cells, leading to pancytopenia. Other infiltrative disorders, such as leukemia, lymphoma, or metastatic solid tumors, can also cause bone marrow failure by physically replacing normal hematopoietic cells.
Clinical Guidelines: A bone marrow biopsy is essential to confirm the diagnosis by demonstrating the presence of fibrosis or other infiltrating cells. A bone marrow aspiration often results in a “dry tap” due to the inability to aspirate liquid marrow. Genetic testing for mutations, such as JAK2, CALR, and MPL, is crucial for diagnosing primary myelofibrosis.
Other Conditions
- Paroxysmal Nocturnal Hemoglobinuria (PNH): This is a rare, acquired clonal disorder of hematopoietic stem cells caused by a somatic mutation in the PIGA gene. This mutation prevents the synthesis of GPI-anchored proteins, including CD55 and CD59, which normally protect blood cells from complement-mediated destruction. PNH is frequently associated with or can evolve from aplastic anemia.
- Large Granular Lymphocyte (LGL) Leukemia: This condition is characterized by a clonal proliferation of cytotoxic T-cells or Natural Killer (NK) cells. These cells produce inhibitory cytokines that suppress hematopoiesis, leading to cytopenias, most commonly neutropenia and anemia.
Inherited Causes of Bone Marrow Failure
Inherited bone marrow failure (BMF) syndromes are a result of genetic mutations that impair HSC function or maintenance. While individually rare, recognizing these conditions is critical for appropriate genetic counseling and tailored management.
Fanconi Anemia (FA)
Pathophysiology: FA is a complex and genetically heterogeneous disorder caused by mutations in one of at least 22 known FAN genes. These genes are crucial for the Fanconi Anemia pathway, which is responsible for DNA repair, specifically interstrand crosslink (ICL) repair. When this pathway is defective, cells accumulate unrepaired DNA damage, leading to chromosome instability, increased apoptosis of HSCs, and progressive bone marrow failure (BMF).
Clinical Guidelines: FA is characterized by congenital malformations (e.g., thumb abnormalities, short stature, café-au-lait spots), developmental delays, and a high predisposition to solid tumors and hematologic malignancies, particularly AML. Diagnosis is confirmed by a chromosomal breakage test (using a DNA crosslinking agent like DEB or MMC) and gene sequencing. Allogeneic HSCT is the only curative treatment for the hematologic manifestations.
Telomere Biology Disorders (TBDs)
Pathophysiology: TBDs, such as dyskeratosis congenita (DC), are caused by mutations in genes (TERT, TERC, DKC1, etc.) that are essential for maintaining telomere length. Telomeres are protective caps at the ends of chromosomes. With each cell division, telomeres shorten. The telomerase enzyme complex is responsible for adding telomeric DNA to prevent critical shortening. In TBDs, defective telomerase leads to accelerated telomere attrition, premature HSC senescence, and bone marrow failure (BMF).
Clinical Guidelines: DC is the most recognized TBD and presents with a classic triad of skin pigmentation, nail dystrophy, and oral leukoplakia. However, bone marrow failure (BMF) is the most life-threatening complication. Diagnosis is based on clinical features, genetic testing, and measurement of telomere length, which is often severely shortened. Treatment options are limited to androgens or allogeneic HSCT.
Other Inherited Syndromes
- Diamond-Blackfan Anemia (DBA): An inherited bone marrow failure (BMF) syndrome characterized by a pure red cell aplasia, often caused by mutations in genes encoding ribosomal proteins. It presents in infancy with severe macrocytic anemia.
- Shwachman-Diamond Syndrome (SDS): An autosomal recessive disorder caused by mutations in the SBDS gene, leading to pancreatic insufficiency, skeletal abnormalities, and bone marrow dysfunction, most commonly neutropenia.
- Severe Congenital Neutropenia (SCN): A heterogeneous group of disorders with a profound absence of neutrophils, often due to mutations in genes like ELANE.
Clinical Approach to Diagnosing Bone Marrow Failure
The diagnostic workup for a patient with suspected bone marrow failure (BMF) is methodical and comprehensive.
- Thorough Clinical History: Detailed patient and family history is critical to identify potential exposures (medications, toxins) and to screen for features of inherited syndromes (congenital abnormalities, family history of hematologic disorders).
- Physical Examination: Looking for signs of pancytopenia.
- Symptoms of Anemia (Low Red Blood Cells): Common symptoms include fatigue, weakness, shortness of breath on exertion (dyspnea), dizziness, headaches, and a pale appearance (pallor) of the skin and mucous membranes. In severe cases, patients may experience chest pain or heart palpitations as the heart works harder to compensate for the lack of oxygen.
- Symptoms of Leukopenia/Neutropenia (Low White Blood Cells): Patients are prone to frequent, recurrent, or severe infections, especially bacterial and fungal infections. The most common signs are fever of unknown origin and mouth sores (oral ulcers), but infections can affect any part of the body, including the lungs (pneumonia), urinary tract, and skin.
- Symptoms of Thrombocytopenia (Low Platelets): Symptoms can range from minor to life-threatening. Patients may notice easy bruising (ecchymoses), tiny, pinpoint red or purple spots on the skin (petechiae), or larger purplish patches (purpura). Spontaneous bleeding from the gums (gingival bleeding) or nosebleeds (epistaxis) is common. In severe cases, bleeding may occur in the gastrointestinal tract (hematemesis, melena) or even the brain (intracranial hemorrhage), which can be fatal.
- Laboratory Studies: A complete blood count (CBC) with differential, reticulocyte count, and peripheral blood smear review.
- Bone Marrow Aspiration and Biopsy: The gold standard for diagnosis. It confirms hypocellularity and helps exclude other causes like leukemia or lymphoma. Morphological assessment, cytogenetics, and molecular testing are essential.
- Specialized Testing: Genetic sequencing for inherited syndromes and flow cytometry for PNH clones are often performed to confirm the underlying etiology.
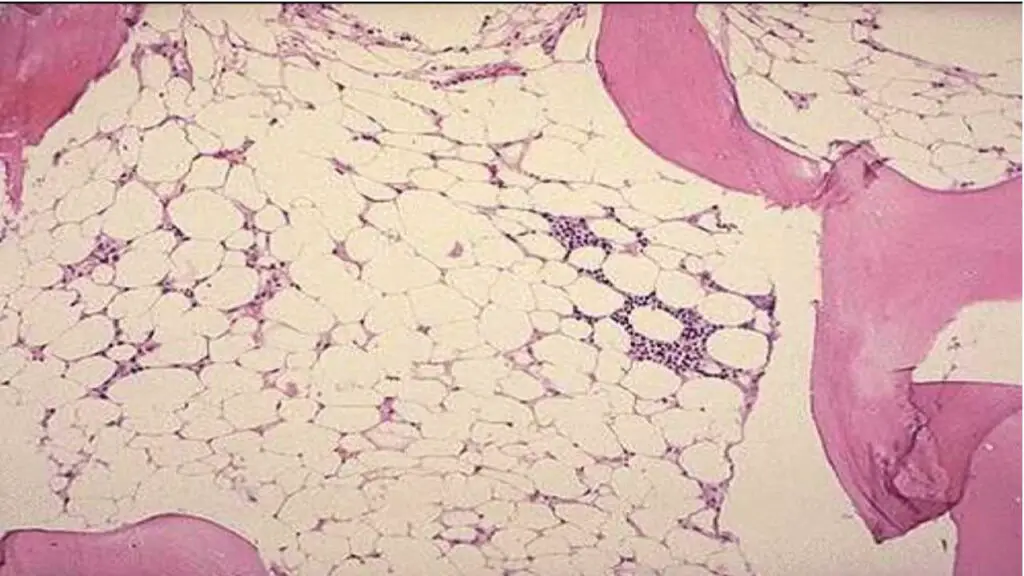
Treatment and Management of Bone Marrow Failure
The treatment of bone marrow failure is multifaceted and depends heavily on the underlying cause, disease severity, and the patient’s age and overall health. It generally involves both supportive care to manage symptoms and definitive treatment to address the root cause.
Supportive Care
This is the cornerstone of management for all patients, regardless of the underlying etiology.
- Blood Transfusions: Packed red blood cell transfusions are given to raise hemoglobin levels and alleviate symptoms of tissue hypoxia. Platelet transfusions are administered to prevent or control bleeding in patients with severe thrombocytopenia.
- Growth Factor Support: Granulocyte Colony-Stimulating Factor (G-CSF) is commonly used to increase neutrophil counts and reduce the risk of severe bacterial infections. Erythropoietin (EPO) may be used to stimulate red blood cell production, although it is typically less effective in aplastic anemia due to the lack of erythroid progenitors.
- Infection Prophylaxis: Patients with severe neutropenia may be given prophylactic antibiotics or antifungals. Strict infection control measures, such as hand hygiene and avoiding exposure to sick individuals, are also crucial.
Definitive Treatment
These therapies aim to cure the disease or modify its progression.
- Immunosuppressive Therapy (IST): The primary treatment for severe acquired aplastic anemia, which is believed to be immune-mediated. The most common regimen is a combination of anti-thymocyte globulin (ATG) and cyclosporine. ATG is an antibody that targets and depletes T-lymphocytes, while cyclosporine is an immunosuppressant that inhibits T-cell activation. The goal is to suppress the autoimmune attack on the hematopoietic stem cells, allowing the bone marrow to recover.
- Hematopoietic Stem Cell Transplantation (HSCT): The only curative treatment for many forms of bone marrow failure, including severe aplastic anemia and certain types of MDS. Allogeneic HSCT, which uses stem cells from a matched donor (preferably a matched sibling), is the standard of care for young patients with a severe disease. The procedure involves conditioning the patient with chemotherapy or radiation to ablate their bone marrow, followed by infusion of the donor’s stem cells. The new stem cells then engraft and begin to produce healthy blood cells, reconstituting the patient’s hematopoietic system.
Management of Underlying Causes
If the cause is a drug or toxin, the offending agent must be immediately withdrawn. For viral-induced bone marrow failure, antiviral therapy may be effective. For inherited disorders, the management often focuses on supportive care and preparing for HSCT, though some conditions may respond to specific agents like androgens in certain cases of Fanconi Anemia.
Conclusion: A Collaborative Approach to Care
The field of bone marrow failure is complex, bridging hematology, genetics, and immunology. A deep understanding of the diverse etiologies and pathophysiological mechanisms is vital for allied health professionals. From the lab technician processing a bone marrow biopsy to the nurse administering a complex drug regimen, each team member plays a critical role in providing comprehensive and compassionate care to patients with these challenging conditions. Continued research into the molecular basis of BMF holds the promise of developing more targeted and effective therapies in the future.
Frequently Asked Questions (FAQs)
What is the main difference between acquired and inherited bone marrow failure?
Acquired bone marrow failure is caused by factors outside of genetics, such as an autoimmune attack or exposure to toxins. Inherited bone marrow failure is caused by genetic mutations present from birth that impair the bone marrow’s function.
Can bone marrow failure be cured?
The potential for a cure depends on the specific cause. For some inherited and acquired conditions, a hematopoietic stem cell transplant (HSCT) can be curative. For others, treatment focuses on managing symptoms and preventing complications.
Are aplastic anemia and myelodysplastic syndrome the same?
No. While both cause pancytopenia, aplastic anemia is a hypoplastic condition with an empty bone marrow, whereas MDS is a hyper- or normocellular condition with ineffective blood cell production and a risk of progression to leukemia.
What are the primary warning signs of bone marrow failure?
The symptoms are related to the lack of blood cells: anemia (fatigue, shortness of breath), neutropenia (frequent infections), and thrombocytopenia (easy bruising, bleeding).
How long can someone live with bone marrow failure?
Glossary of Medical Terms
- Aplastic Anemia: A type of bone marrow failure where the marrow is severely hypocellular, leading to a deficiency of all three blood cell types.
- Cytopenia: A deficiency of one or more types of blood cells.
- Hematopoietic Stem Cells (HSCs): Multipotent stem cells located in the bone marrow that give rise to all types of mature blood cells.
- Hypocellularity: A state of having fewer cells than normal, as seen in a bone marrow biopsy of an aplastic patient.
- Myelodysplastic Syndromes (MDS): A group of clonal disorders of the hematopoietic stem cells, characterized by ineffective blood production and an increased risk of acute leukemia.
- Pancytopenia: A severe deficiency of all three major blood cell types: red blood cells, white blood cells, and platelets.
- Telomeres: Protective DNA caps at the ends of chromosomes that shorten with each cell division.
- Fanconi Anemia: An inherited DNA repair disorder that leads to progressive bone marrow failure.
Disclaimer: This article is intended for informational purposes only and is specifically targeted towards medical students. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Moore CA, Krishnan K. Bone Marrow Failure. [Updated 2023 Jul 10]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK459249/
- Fox, L. C., McQuilten, Z. K., Firkin, F., Fox, V., Badoux, X., Bajel, A., Barbaro, P., Cole-Sinclair, M. F., Forsyth, C., Gibson, J., Hiwase, D. K., Johnston, A., Mills, A., Roncolato, F., Sutherland, R., Szer, J., Ting, S. B., Vilcassim, S., Young, L., Waters, N. A., … Australian Aplastic Anaemia and other Bone Marrow Failure Syndromes Registry (AAR) Investigators (2023). The Australian Aplastic Anaemia and other Bone Marrow Failure Syndromes Registry. Best practice & research. Clinical haematology, 36(4), 101516. https://doi.org/10.1016/j.beha.2023.101516
- Iwafuchi H. (2018). The histopathology of bone marrow failure in children. Journal of clinical and experimental hematopathology : JCEH, 58(2), 68–86. https://doi.org/10.3960/jslrt.18018
- Collins, J., & Dokal, I. (2015). Inherited bone marrow failure syndromes. Hematology (Amsterdam, Netherlands), 20(7), 433–434. https://doi.org/10.1179/1024533215Z.000000000381
- Deng, J., & McReynolds, L. J. (2023). Inherited bone marrow failure syndromes: a review of current practices and potential future research directions. Current opinion in pediatrics, 35(1), 75–83. https://doi.org/10.1097/MOP.0000000000001196
- Groarke, E. M., Young, N. S., & Calvo, K. R. (2021). Distinguishing constitutional from acquired bone marrow failure in the hematology clinic. Best practice & research. Clinical haematology, 34(2), 101275. https://doi.org/10.1016/j.beha.2021.101275
- Pasca, S., & Gondek, L. P. (2021). Clonal hematopoiesis and bone marrow failure syndromes. Best practice & research. Clinical haematology, 34(2), 101273. https://doi.org/10.1016/j.beha.2021.101273
- Malouf, C., Loughran, S. J., Wilkinson, A. C., Shimamura, A., & Río, P. (2022). Translational research for bone marrow failure patients. Experimental hematology, 105, 18–21. https://doi.org/10.1016/j.exphem.2021.11.004