Key Takeaways
Beta-thalassemia is an inherited disorder caused by mutations in the HBB gene that reduce or stop production of the beta-globin chain of hemoglobin. The resulting imbalance between alpha and beta chains drives ineffective red cell production, ongoing red cell breakdown, and chronic anemia.
- Spectrum of Severity ▾: The disease varies widely, categorized generally by how much medical support is needed. It ranges from the silent, symptom-free "minor" trait, to "intermedia," to the most severe "major" form (Transfusion-Dependent Thalassemia), which requires lifelong blood transfusions starting in infancy.
- The Threat of Iron Overload ▾: For patients who depend on transfusions, iron overload is the dominant long-term health risk. Driven by both the transfused blood itself and increased intestinal absorption, toxic iron levels can accumulate in the heart, liver, and endocrine glands if not managed with iron-removing chelation therapy.
- Investigations & Diagnosis ▾: Diagnosis requires a multi-step approach. It typically starts with a Complete Blood Count (CBC) and blood smear, followed by hemoglobin electrophoresis (to look for raised HbA2 and HbF levels), and is confirmed by DNA testing to pinpoint the specific genetic mutation. Cardiac and liver T2* MRI are now standard for monitoring iron in specific organs.
- Treatments and Prognosis ▾: Life expectancy for severe cases has dramatically improved, with patients now living into their 50s, 60s, and beyond. While standard care involves regular transfusions and iron chelation, modern medicine has introduced advanced options like stem-cell transplants, luspatercept, and curative gene therapies (such as CASGEVY and Zynteglo) offer potential cures for eligible patients.
*Click ▾ for more information
What is beta-thalassemia?
Beta-thalassemia is a genetic disorder where the body makes too little (or none) of the beta chain of hemoglobin [9,11]. Hemoglobin is the protein inside red blood cells that carries oxygen, and it normally contains two alpha chains and two beta chains.
It belongs to the wider thalassemia family. Alpha-thalassemia, its sibling condition, affects the alpha chain instead. In beta-thalassemia, alpha chain production keeps running normally. The unpaired alpha chains then build up inside developing red blood cells, where they clump together, damage cell membranes, and cause two key problems: red cells dying inside the bone marrow before they ever leave it, and ongoing red cell breakdown in the bloodstream.[9,16]
The beta-globin gene and hemoglobin
The HBB gene sits on chromosome 11. Adults carry two copies, one inherited from each parent. Each copy provides instructions for making the beta-globin chain.
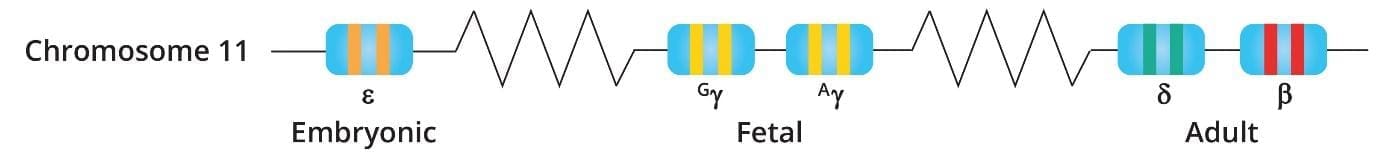
Adult hemoglobin (HbA) is made of two alpha and two beta subunits. Two minor adult forms also exist: HbA2 (1.5–3.5% of total hemoglobin) and HbF, the fetal form (less than 1% in adults). Each hemoglobin molecule can carry up to four oxygen molecules through small iron-containing pockets called heme groups [10,11].
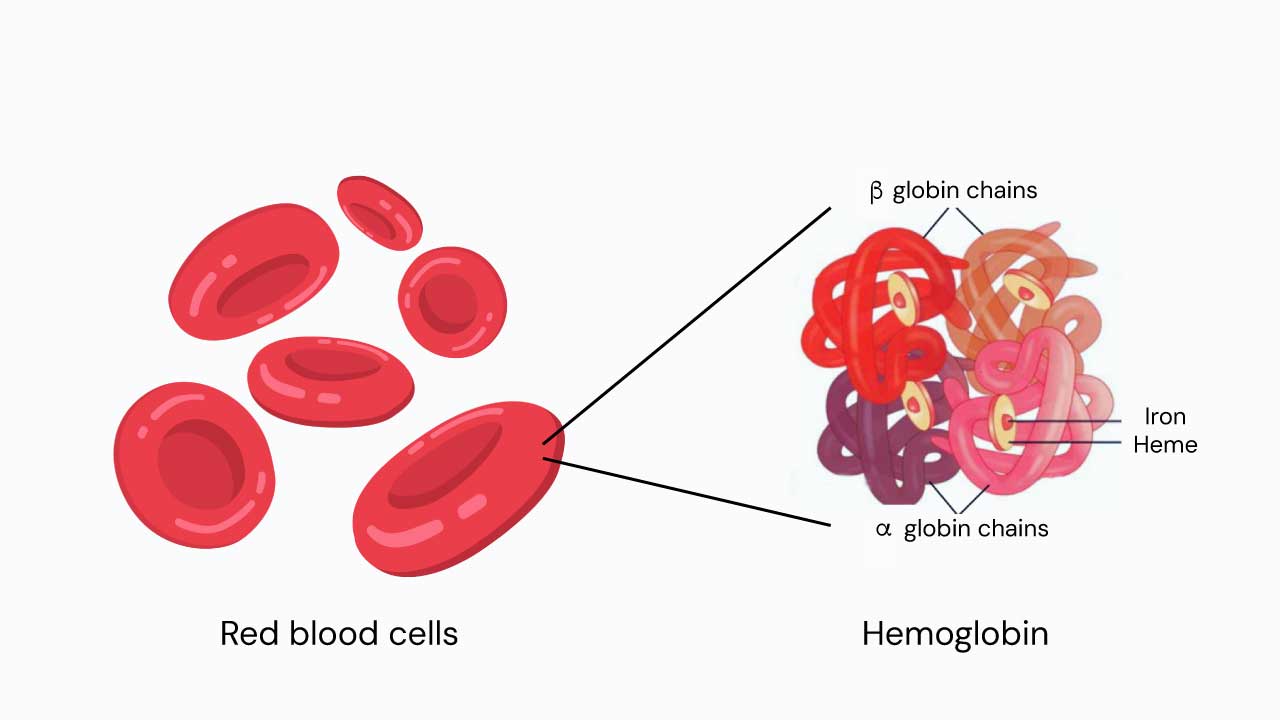
More than 350 different mutations of HBB have been identified. Some reduce beta chain output (called β+ mutations). Others stop it completely (β0 mutations). The exact mutation a person inherits determines how severe their disease will be.[11]
Pathophysiology: how the disease unfolds
Three connected processes drive beta-thalassemia [9,16].
- The chain imbalance. Without enough beta chains, free alpha chains pile up inside young red blood cells. They oxidize, clump, and damage the cells from the inside.
- Ineffective erythropoiesis. This means red blood cells dying in the bone marrow before they finish maturing. The marrow tries to compensate by working harder, expanding into surrounding bone. Over time, this expansion thins the bones, distorts the face and skull, and weakens the skeleton. The body even sets up backup red cell factories in the liver, spleen, and other tissues. This is called extramedullary hematopoiesis (blood cell production outside the bone marrow).
- Hepcidin suppression and iron overload. Stressed red cell precursors release a hormone called erythroferrone (ERFE)[13]. ERFE suppresses hepcidin, the hormone that normally controls how much iron the gut absorbs. With hepcidin turned down, the intestine soaks up far more iron than the body needs.
The Cost of Chronic Transfusions
For patients on regular transfusions, the problem doubles. Each unit of transfused blood delivers around 200–250 mg of extra iron. The body has no way to get rid of this excess. Iron then accumulates in the heart, liver, and endocrine glands, where its most toxic forms — non-transferrin-bound iron (NTBI) and labile plasma iron (LPI) — slowly damage organs.
Inheritance and Epidemiology
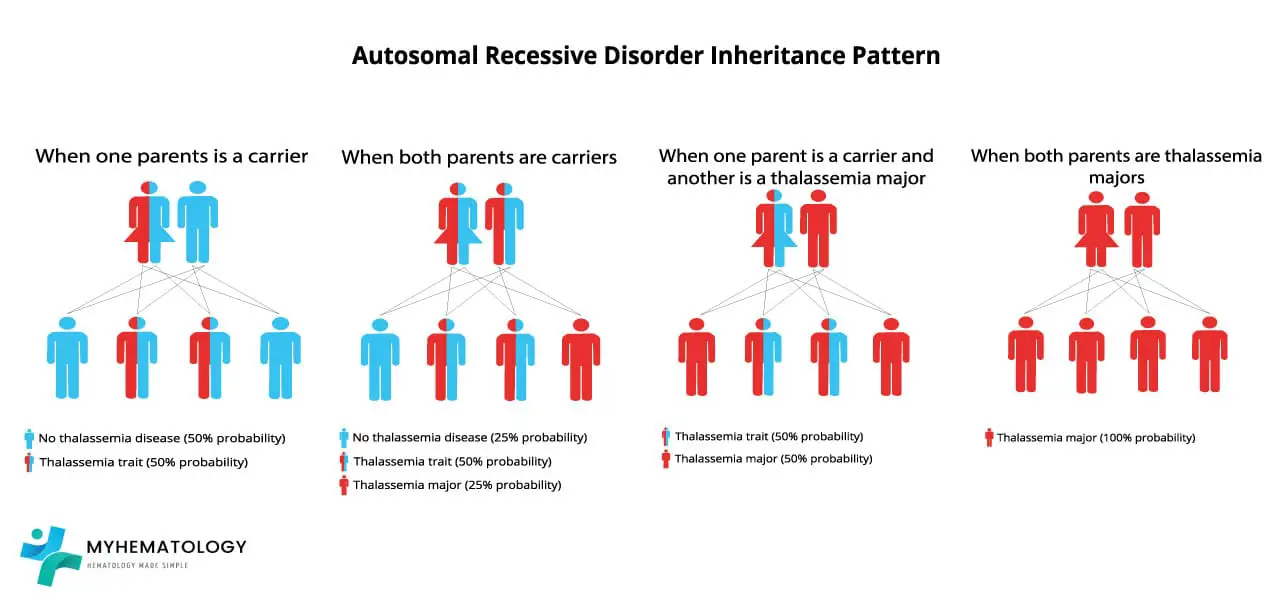
Beta-thalassemia is autosomal recessive. That means a child needs two faulty copies of the HBB gene, one from each parent, to develop the disease.
If both parents are carriers, every pregnancy carries the same odds:
- 25% chance the child has beta-thalassemia major
- 50% chance the child is a carrier (minor)
- 25% chance the child inherits two normal alleles
If only one parent is a carrier, no child will have the disease itself. However, each child still has a 50% chance of being a carrier [11].
Carrier rates reflect the historical reach of malaria, since carrying one thalassemia gene offers some protection against severe malaria. The trait is found in roughly 12–14% of people in Cyprus and Sardinia, and in 3–10% across much of Southeast Asia [12]. It is also common across the Middle East, North Africa, and the Indian subcontinent. Migration has spread these genes worldwide. Many countries now run carrier screening and prenatal diagnosis programs to reduce the number of severely affected births.[1,12]
Molecular Classifications
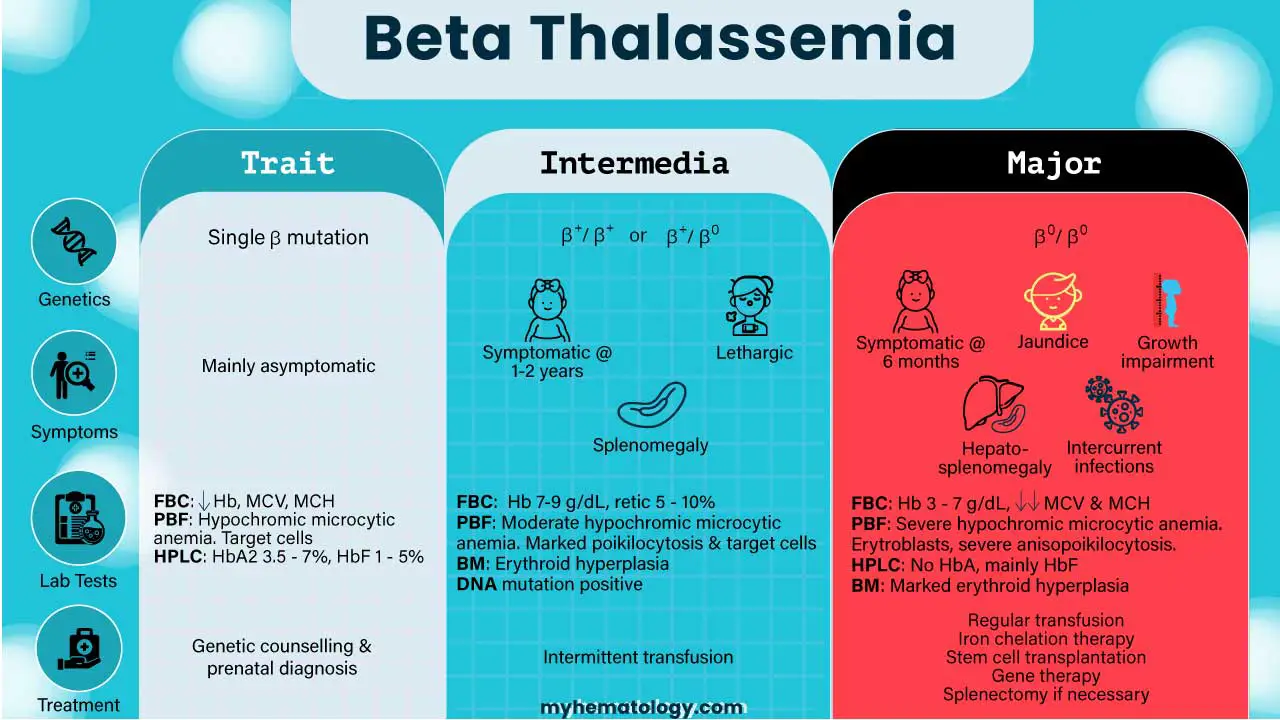
Beta-thalassemia can be loosely classified in two overlapping ways. The first is by genotype:
- Beta thalassemia minor (trait). One mutated allele.
- Beta thalassemia intermedia. Two milder mutations, or two severe mutations softened by other inherited factors such as co-existing alpha-thalassemia or hereditary persistence of fetal hemoglobin (HPFH).
- Beta thalassemia major. Two severe mutations (homozygous or compound heterozygous, usually β0) [9,11].
The second classification is clinical and increasingly preferred: transfusion-dependent thalassemia (TDT) versus non-transfusion-dependent thalassemia (NTDT) [1,3]. This split matters more in practice because it directly guides treatment decisions.
Classification at a glance
Hematology · Classification
| Feature | Mild Beta Thalassemia Minor Trait | Moderate Beta Thalassemia Intermedia | Severe Beta Thalassemia Major Cooley's Anemia |
|---|---|---|---|
| Genotype | One mutated β allele (heterozygote) | Two milder mutations, or severe mutations softened by other inherited factors | Two severe mutations (often β0/β0) |
| Hemoglobin production | Slight drop in β chains; α/β ratio almost balanced | Reduced β chains, modest α-chain excess | Almost no β chains, large α-chain excess |
| Onset | Usually picked up incidentally | Childhood to adulthood (often 2–6 years) | First year of life (usually 3–6 months) |
| Transfusions | Not needed | Sometimes, for stress or illness | Every 2–4 weeks, lifelong |
| Common features | Small red cells, possibly mild anemia | Pallor, enlarged spleen, bone changes, growth delay, clotting risk | Severe anemia, failure to thrive, jaundice, bone deformity, organ enlargement, iron overload |
| Life expectancy with optimal care | Normal | Variable, often near-normal | 50s–60s and rising [2,16] |
Categories shown in order of increasing clinical severity (Mild → Moderate → Severe).
Signs & Symptoms
What a patient experiences depends on which form of beta-thalassemia they have.
Beta thalassemia minor is usually silent. People often discover it by accident when a routine blood test shows small, pale red cells. A raised HbA2 on hemoglobin testing confirms it.
Beta thalassemia intermedia typically appears in childhood or later. Patients may have mild to moderate anemia, pallor, an enlarged spleen, gallstones from chronic red cell breakdown, bone changes, and a higher risk of blood clots and pulmonary hypertension. Clot and pulmonary hypertension risk is actually higher in NTDT than in well-transfused TDT, because the chronic hemolysis (red cell breakdown) is never fully corrected [3].
Beta thalassemia major develops between 3 and 6 months of age. As fetal hemoglobin naturally declines after birth, the child cannot replace it with normal adult hemoglobin. Symptoms include severe anemia, failure to thrive, jaundice, and the classic "thalassemia facies" with frontal bossing and overgrowth of the upper jaw. Without transfusion, most affected children die in the first decade of life [1,11]
Complications
In treated transfusion-dependent thalassemia, iron overload is the dominant long-term threat. It causes a cascade of organ damage [1,2]:
- Heart. Cardiomyopathy (weakened heart muscle), heart failure, and abnormal heart rhythms. Historically, this was the leading cause of death.
- Liver. Fibrosis, cirrhosis, and an increased risk of liver cancer, especially when viral hepatitis is also present.
- Endocrine glands. Delayed or absent puberty, short stature, underactive thyroid, low parathyroid function, and diabetes mellitus.
- Bones. Osteoporosis and fractures.
Other complications include thrombosis (especially after splenectomy and in NTDT), pulmonary hypertension, gallstones, leg ulcers, and infections — particularly in patients whose spleen has been removed [3,15].
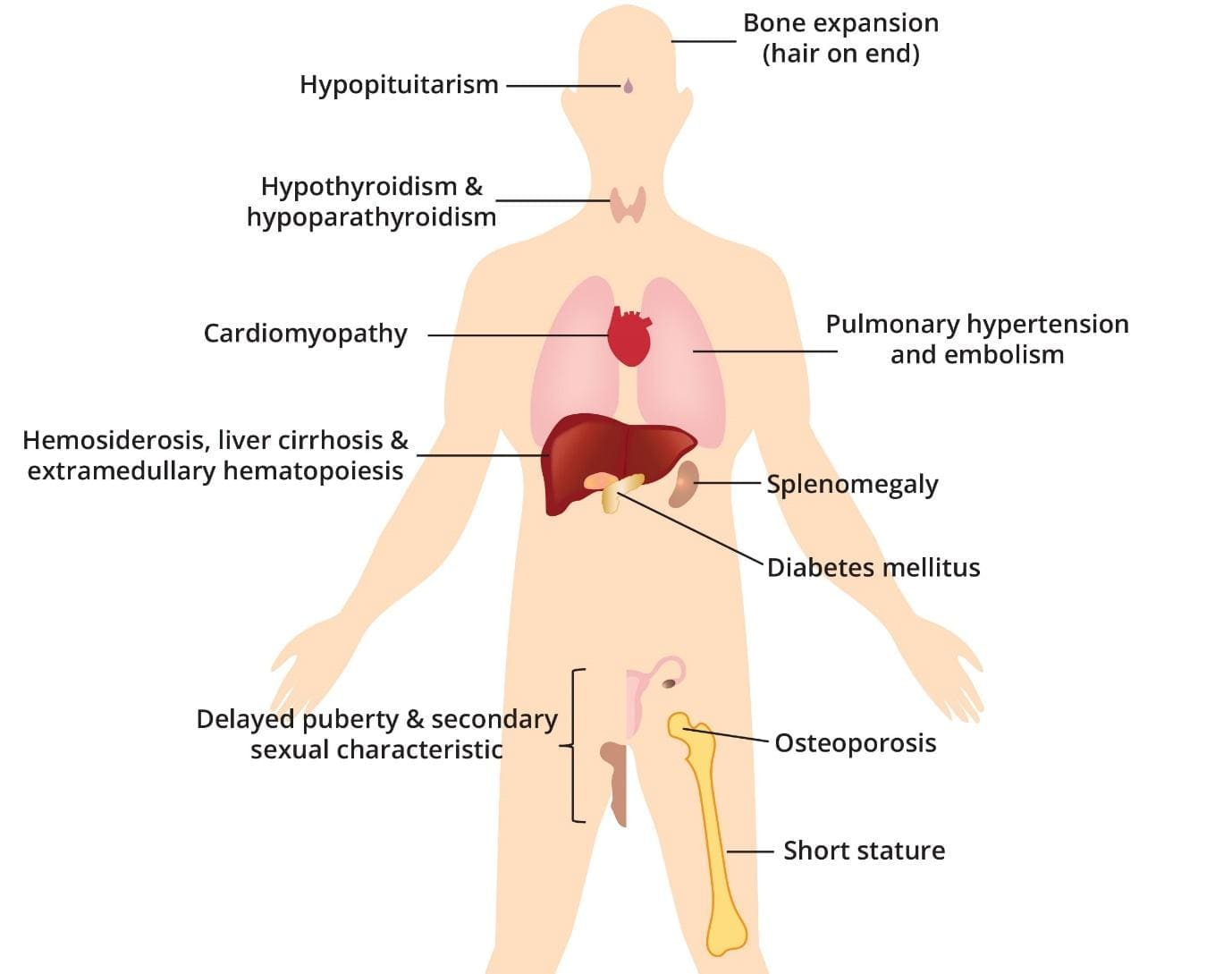
Beta-thalassemia major results in the abnormal production of hemoglobin, leading to chronic anemia. The repeated blood transfusions required to manage anemia introduce excess iron into the body, gradually accumulating in various organs.
Laboratory investigations
Testing for beta-thalassemia happens in layers, from cheap screening to definitive genetic confirmation.
Complete blood count (CBC)
The first clue. Look for low hemoglobin, low MCV (mean corpuscular volume, the size of red cells, often under 70 fL), and low MCH (mean corpuscular hemoglobin, the amount of hemoglobin per cell). The reticulocyte count (young red cells) rises in intermedia and major.
RBC Count: Thalassemia Trait vs. IDA
In thal trait, the red cell count is often paradoxically high. This helps distinguish trait from iron deficiency, which usually shows a low red cell count.
Peripheral blood smear
Trait shows mild microcytosis with target cells. Intermedia shows moderate small, pale red cells with raised reticulocytes. Major shows striking variation in red cell size and shape, target cells, basophilic stippling (small dots inside red cells), and nucleated red cells.
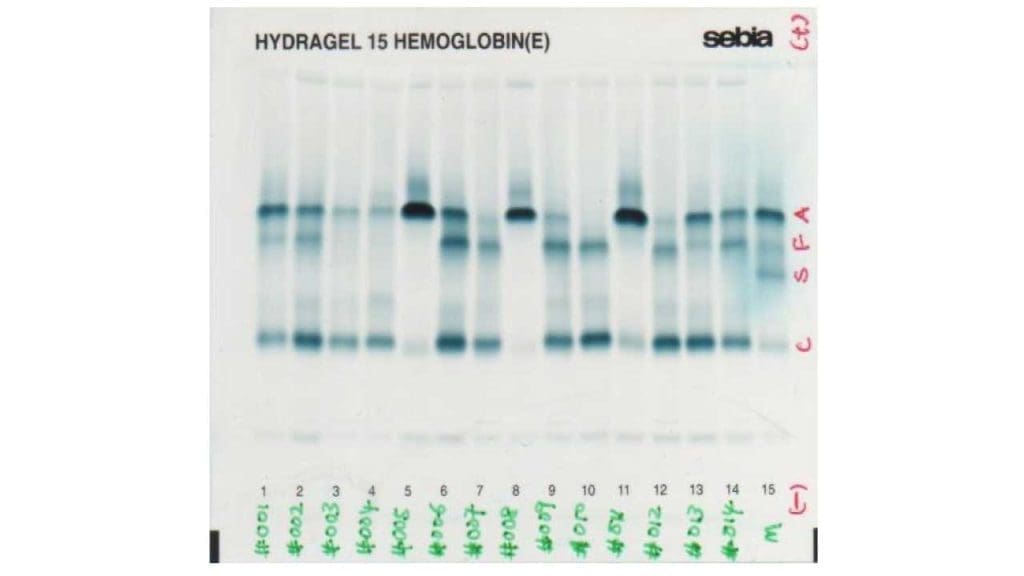
Hemoglobin HPLC and Electrophoresis
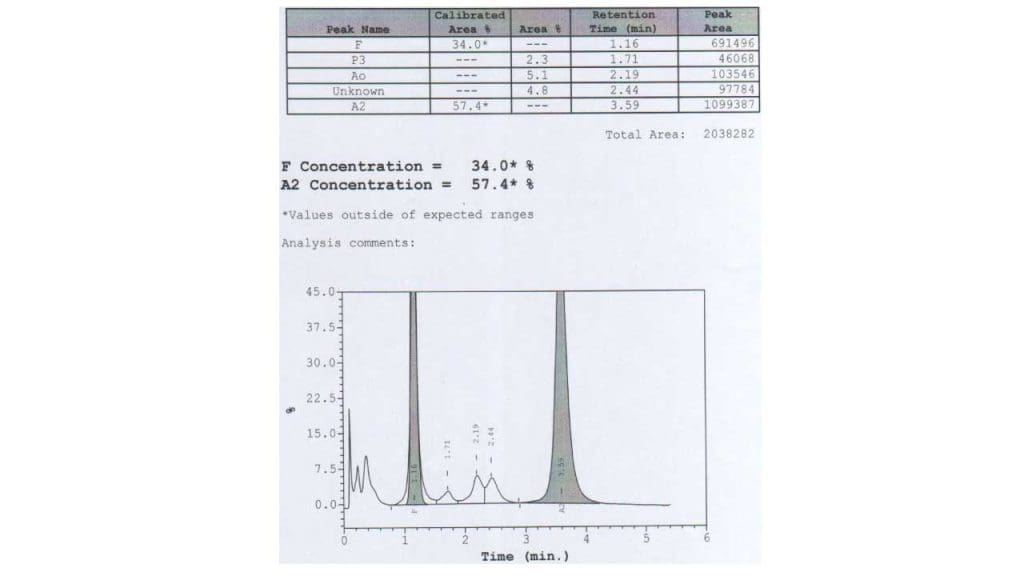
These tests separate the different types of hemoglobin and quantify each [10,11]. In trait, HbA2 is raised above 3.5%, often with a slight rise in HbF. In major, HbA is barely present or absent, HbF dominates (often above 90%), and HbA2 is also raised.
Recent 2025–2026 updates to global gynecological and obstetric guidelines have further cemented High-Performance Liquid Chromatography (HPLC) as the absolute "gold standard" for carrier screening. Guidelines now strongly emphasize performing HPLC early in a pregnancy (preferably before 8 weeks gestation) rather than relying on standard complete blood counts, to ensure at-risk couples have adequate time for genetic counseling [20,21].
Caution
On most HPLC systems, HbE elutes (comes off the column) at the same point as HbA2. In someone with both HbE and beta-thalassemia, the combined peak can exceed 80%. This is not "very high HbA2" — it is mostly HbE.
DNA testing
Direct sequencing or targeted PCR pinpoints the exact HBB mutation. This is essential for prenatal diagnosis, preimplantation genetic testing, gene therapy eligibility, and clarifying compound heterozygotes [11]
Iron studies and organ-iron monitoring
Serum ferritin, transferrin saturation, and serum iron all rise with iron overload. TIBC (total iron-binding capacity) is usually normal to low. Ferritin alone is not enough, though. The standard of care now uses cardiac T2* MRI and liver iron concentration (LIC) MRI to measure iron in specific organs [14]. Treatment goals are clear: ferritin under 1000 ng/mL, cardiac T2* above 20 ms, and LIC below 7 mg of iron per gram of dry liver tissue [1,2].
Other investigations
Bilirubin and urobilinogen rise (signs of red cell breakdown). Haptoglobin drops. Routine surveillance also covers liver function, fasting glucose or oral glucose tolerance testing (annually from age 10), thyroid and parathyroid hormones, vitamin D, bone density (DEXA), echocardiogram, sex hormones, and screening for infections that can spread through transfusion.[1,2]
Treatment and Management
Treatment has three goals: correct the anemia, prevent and treat iron overload, and catch complications early. The right approach depends on the type of beta-thalassemia.
Beta thalassemia minor
No specific treatment is needed. Genetic counseling matters most, especially before starting a family. Iron supplements should not be given unless iron deficiency has been confirmed separately. Giving iron to a thalassemia carrier who is not iron-deficient can quietly add to iron overload.
Beta thalassemia intermedia (NTDT)
Most patients manage without regular transfusions. They may need them during illness, pregnancy, growth spurts, or surgery. Hydroxyurea can boost fetal hemoglobin in selected patients and ease symptoms. Iron chelation is often still needed because hepcidin suppression keeps the gut absorbing too much iron, even without transfusions [3].
Beta thalassemia major (TDT)
This is the most demanding form to manage, and treatment runs lifelong.
Regular red cell transfusions form the backbone of care. Patients receive packed red cells every 2–4 weeks. The aim is to keep the pre-transfusion hemoglobin between 9.5 and 10.5 g/dL [1,2]. At this level, the body stops trying to produce its own faulty red cells, which prevents bone deformity and supports normal growth.
Iron chelation therapy starts after about 10–20 transfusions, or once ferritin climbs above roughly 1000 ng/mL. Three medications are licensed [1,2,15]:
- Deferoxamine (Desferal). Given as a slow injection under the skin over 8–12 hours, 5–7 nights a week.
- Deferasirox (Exjade, Jadenu). Once-daily tablet or dispersible.
- Deferiprone (Ferriprox). Three times daily by mouth. Particularly good at reaching iron in the heart.
For patients with severe cardiac iron loading, doctors often combine deferiprone with deferoxamine. The choice is tailored to each patient using MRI results, ferritin trends, side effects, and how easily the patient can stick with the regimen.
Luspatercept (Reblozyl) is a newer injectable drug that helps red cell precursors finish maturing properly [4]. It works by blocking signals (specifically in the Smad2/3 pathway) that normally hold back late-stage red cell development. In the phase 3 BELIEVE trial in adult TDT patients, 21.4% of luspatercept-treated patients achieved a reduction of 33% or more in transfusion burden during weeks 13 to 24, compared with 4.5% on placebo (P<0.001) [4]. In long-term follow-up, around 77% of patients sustained this kind of transfusion reduction across any 12-week interval over a median of three years [5]. It is given by injection under the skin every three weeks.
Mitapivat (Aqvesme) is a groundbreaking, first-in-class oral medication recently approved for the treatment of anemia in adults with both transfusion-dependent and non-transfusion-dependent alpha- and beta-thalassemia. It works as a pyruvate kinase (PK) activator, directly boosting cellular energy (ATP) production within red blood cells to enhance their survival and significantly reduce their premature destruction (hemolysis). By fundamentally modifying beta-thalassemia disease process, the drug greatly decreases the reliance on lifelong blood transfusions for severe cases while notably improving hemoglobin levels and reducing fatigue in milder forms. However, because the medication carries a risk of liver toxicity, its use requires strict, routine liver function monitoring through a mandatory safety program, especially during the first 24 weeks of treatment [16,17,18,19].
Mandatory Liver Function Testing (REMS Program)
Mitapivat (Aqvesme) carries a Boxed Warning for potential hepatocellular injury (liver toxicity). Consequently, new investigation protocols are legally required in the U.S. under a Risk Evaluation and Mitigation Strategy (REMS) program. Patients must undergo baseline liver function tests before their first dose, followed by routine blood tests every four weeks for the first 24 weeks of treatment, and then as clinically indicated.
Allogeneic hematopoietic stem-cell transplantation (HSCT) — replacing the patient's bone marrow with healthy donor stem cells — remains the most established curative option. It works best in young, well-chelated patients with an HLA-matched sibling donor [1].
Gene therapy has reshaped what "cure" can mean for beta-thalassemia:
- Betibeglogene autotemcel (Zynteglo). Adds a working β-globin gene using a lentiviral vector. FDA-approved in August 2022 for both adult and pediatric TDT [8]
- Exagamglogene autotemcel (CASGEVY). Uses CRISPR/Cas9 to edit the BCL11A gene's red cell enhancer, switching fetal hemoglobin back on. FDA-approved for TDT on 16 January 2024 for patients aged 12 and older [7]. In the pivotal trial, most evaluable TDT patients became transfusion-independent for at least 12 months in a row [7]
Splenectomy is now used much less than in past decades. Long-term risks of clotting, infection, and pulmonary hypertension have shifted the balance. When it cannot be avoided, surgeons usually wait until age 5 or 6, give all encapsulated-organism vaccines (pneumococcal, meningococcal, Haemophilus influenzae type b) beforehand, and start antibiotic prophylaxis afterward [1].
Supportive care and surveillance
Several extras matter alongside core treatment:
- Folic acid (1–5 mg daily) supports the high red cell turnover.
- Iron supplements should be avoided unless iron deficiency is independently confirmed.
- Vaccinations follow the standard schedule, with extra cover for encapsulated organisms after splenectomy.
- Annual surveillance includes cardiac T2* MRI, liver T2*/LIC MRI, ferritin trend, oral glucose tolerance test, thyroid and parathyroid function, bone density scan, sex hormones, growth monitoring, and infection screening [1,2].
Caregiver and patient considerations
For families, beta-thalassemia is a daily reality, not just a diagnosis. Transfusion days take 3–4 hours every 2–4 weeks. Chelation must be taken every day, often for life. Adherence to chelation is the strongest predictor of long-term outcome. More important than any other single factor.
Practical issues include school accommodations on transfusion days, careful planning for the move from pediatric to adult care in the late teens, and ongoing emotional and psychological support. The Thalassaemia International Federation publishes patient-facing resources and supports more than 200 national patient associations worldwide [1].
For couples where one or both partners are carriers, preconception genetic counseling is essential. Preimplantation genetic testing and chorionic villus sampling between 10 and 13 weeks of pregnancy are widely available reproductive options.
Prognosis
The outlook for beta-thalassemia has changed enormously in a generation. With optimal modern care, life expectancy in TDT now reaches into the 50s and 60s, and continues to climb [2,16]. The clinical concerns have shifted too — from childhood survival to adult-onset complications like diabetes, heart disease, osteoporosis, and secondary cancers.
In well-resourced settings, gene therapy is pushing the curative ceiling higher still. Globally, however, access to safe transfusion, MRI-based iron monitoring, and consistent chelation remains very uneven. The Thalassaemia International Federation continues to advocate for equitable care worldwide [1,2].
Frequently Asked Questions (FAQs)
What is the life expectancy of a person with beta-thalassemia?
Beta-thalassemia trait carriers have a normal life expectancy. Intermedia varies widely depending on genotype and quality of care. Major now reaches the 50s and 60s with optimal transfusion and chelation, and may extend further with gene therapy [2,16].
Does beta-thalassemia get worse with age?
The genetic defect itself does not change. However, the cumulative effect of iron loading makes complications such as heart disease, liver damage, hormone problems, and osteoporosis more likely over time. Consistent chelation and modern monitoring delay or prevent most of these [1,2].
Can people with thalassemia donate blood?
People with intermedia and major depend on transfusions and cannot donate. People with thalassemia trait may be accepted or deferred depending on local blood-service rules, hemoglobin level, and iron status. Always check with the local blood service.
How is beta-thalassemia diagnosed?
Beta-thalassemia is diagnosed through a series of investigations. A complete blood count and blood smear point toward small, pale red cells. Hemoglobin HPLC or electrophoresis showing raised HbA2 (above 3.5%) confirms the trait. The HbF and HbA pattern characterizes intermedia and major. DNA testing of HBB identifies the exact mutation, which is essential for prenatal counseling and gene therapy eligibility [10,11].
Is gene therapy a cure?
For many transfusion-dependent beta-thalassemia patients, betibeglogene autotemcel and exagamglogene autotemcel produce lasting transfusion independence and amount to a functional cure. Limitations include high cost, the need for myeloablative chemotherapy beforehand, an age limit (12 years and older for CASGEVY), and the need for specialized centers.[7,8]
Why is iron overload a problem even in non-transfused patients?
Ineffective red cell production releases erythroferrone, which suppresses hepcidin. With hepcidin turned down, the gut absorbs too much iron. NTDT patients can therefore develop significant iron overload without ever being transfused, which is why chelation is sometimes still needed [3,13].
Glossary of Related Medical Terms
- Allele — One of two or more alternative versions of a gene at the same chromosomal location. A person inherits one allele from each parent.
- Anemia — A condition where the blood has too little hemoglobin or too few healthy red blood cells, reducing oxygen delivery to tissues.
- Anisopoikilocytosis — Variation in both size (anisocytosis) and shape (poikilocytosis) of red blood cells seen on a blood smear.
- Autosomal recessive inheritance — A pattern in which a person needs two faulty copies of a gene (one from each parent) to develop the condition; one copy makes them a carrier without illness.
- β-globin gene (HBB) — The gene on chromosome 11 that codes for the beta-globin protein chain in adult hemoglobin.
- BCL11A — A gene that normally switches off fetal hemoglobin production after birth. Disabling its erythroid enhancer reactivates HbF — the basis of CASGEVY gene therapy.
- Chelation therapy — Treatment with drugs that bind excess iron in the body so it can be excreted in urine or stool.
- Compound heterozygosity — Inheriting two different mutations of the same gene — one from each parent — rather than two copies of the same mutation.
- Cooley's anemia — Historical name for beta thalassemia major.
- CRISPR/Cas9 — A precise gene-editing tool that cuts DNA at a chosen location, allowing the body's repair machinery to disable or modify a target gene.
- Erythroferrone (ERFE) — A hormone released by red-cell precursors that suppresses hepcidin, increasing intestinal iron absorption.
- Erythropoiesis — The production of red blood cells in the bone marrow.
- Extramedullary hematopoiesis — Blood-cell production outside the bone marrow, typically in the liver and spleen, when marrow cannot meet demand.
- Fetal hemoglobin (HbF) — The dominant hemoglobin before birth (α2γ2). Reactivating HbF in adults compensates for missing beta chains.
- Hb A — The main adult hemoglobin (α2β2), about 95–98% of adult hemoglobin normally.
- Hb A2 — A minor adult hemoglobin (α2δ2), normally 1.5–3.5%; raised (>3.5%) in beta thalassemia trait.
- Hemoglobin electrophoresis — A laboratory test that separates hemoglobin types by electrical charge to identify abnormal variants.
- Hemolysis — Premature destruction of red blood cells.
- Hepcidin — The master regulator hormone of iron metabolism; when low, the gut absorbs more iron.
- Hepatosplenomegaly — Enlargement of both liver and spleen.
- HPLC (High-Performance Liquid Chromatography) — Automated technique that separates and quantifies hemoglobin variants; the modern alternative to gel electrophoresis.
- Ineffective erythropoiesis — Premature death of red-cell precursors in the bone marrow before they mature.
- Iron overload (hemosiderosis) — Accumulation of excess iron in body tissues, damaging the heart, liver, and endocrine glands.
- Labile plasma iron (LPI) — A reactive, toxic form of circulating iron that drives organ damage in iron overload.
- Liver iron concentration (LIC) — Iron content per gram of dry liver tissue (mg Fe/g dw); measured non-invasively by MRI.
- Luspatercept — An injectable medicine that helps red-cell precursors mature, reducing transfusion needs in adults with TDT.
- MCH (Mean Corpuscular Hemoglobin) — Average mass of hemoglobin per red blood cell; low in thalassemia.
- MCV (Mean Corpuscular Volume) — Average size of red blood cells; low (microcytic) in thalassemia.
- Microcytic, hypochromic — Red cells that are smaller than normal and paler than normal.
- Non-transfusion-dependent thalassemia (NTDT) — Forms of thalassemia where regular lifelong transfusions are not required for survival.
- Pretransfusion hemoglobin — The Hb level measured immediately before a transfusion; the target in TDT is typically 9.5–10.5 g/dL.
- Reticulocyte — A young red blood cell newly released from bone marrow; raised when the marrow is trying to compensate for anemia.
- Splenectomy — Surgical removal of the spleen.
- T2* MRI — A specialized magnetic-resonance imaging sequence that quantifies iron loading in the heart and liver.
- Target cell — A red cell with a central spot of hemoglobin resembling a target; common in thalassemia and other hemoglobinopathies.
- Transferrin saturation — The percentage of transferrin (the iron-transport protein) that is bound to iron; raised in iron overload.
- Transfusion-dependent thalassemia (TDT) — Severe thalassemia requiring regular lifelong red-cell transfusions to survive.
Disclaimer: This article is intended for educational and informational purposes only. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Taher AT, Farmakis D, Porter JB, et al., editors. Guidelines for the Management of Transfusion-Dependent β-Thalassaemia (TDT) [Internet]. 5th edition. Nicosia, Cyprus: Thalassaemia International Federation; 2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK614251/
- Musallam, K. M., Cappellini, M. D., Porter, J. B., Farmakis, D., Eleftheriou, A., Angastiniotis, M., & Taher, A. T. (2025). TIF Guidelines for the Management of Transfusion-Dependent β-Thalassemia. HemaSphere, 9(3), e70095. https://doi.org/10.1002/hem3.70095
- Taher AT, Musallam KM, Cappellini MD. Guidelines for the Management of Non-Transfusion-Dependent β-Thalassaemia [Internet]. 3rd edition. Nicosia (Cyprus): Thalassaemia International Federation; 2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK599489/
- Cappellini, M. D., Viprakasit, V., Taher, A. T., Georgiev, P., Kuo, K. H. M., Coates, T., Voskaridou, E., Liew, H. K., Pazgal-Kobrowski, I., Forni, G. L., Perrotta, S., Khelif, A., Lal, A., Kattamis, A., Vlachaki, E., Origa, R., Aydinok, Y., Bejaoui, M., Ho, P. J., Chew, L. P., … BELIEVE Investigators (2020). A Phase 3 Trial of Luspatercept in Patients with Transfusion-Dependent β-Thalassemia. The New England journal of medicine, 382(13), 1219–1231. https://doi.org/10.1056/NEJMoa1910182
- Cappellini, M. D., Viprakasit, V., Georgiev, P., Coates, T. D., Origa, R., Khelif, A., Liew, H. K., Tantiworawit, A., Chew, L. P., Khalil, A., Ho, P. J., Kuo, K. H. M., Holot, N., Perin, M., Giuseppi, A. C., Kuo, W. L., Lai, Y., Medlin, L. F., Bueno, L. M., Kattamis, A., … Taher, A. T. (2025). Long-term efficacy and safety of luspatercept for the treatment of anaemia in patients with transfusion-dependent β-thalassaemia (BELIEVE): final results from a phase 3 randomised trial. The Lancet. Haematology, 12(3), e180–e189. https://doi.org/10.1016/S2352-3026(24)00376-4
- Frangoul, H., Locatelli, F., Sharma, A., Bhatia, M., Mapara, M., Molinari, L., Wall, D., Liem, R. I., Telfer, P., Shah, A. J., Cavazzana, M., Corbacioglu, S., Rondelli, D., Meisel, R., Dedeken, L., Lobitz, S., de Montalembert, M., Steinberg, M. H., Walters, M. C., Eckrich, M. J., … CLIMB SCD-121 Study Group (2024). Exagamglogene Autotemcel for Severe Sickle Cell Disease. The New England journal of medicine, 390(18), 1649–1662. https://doi.org/10.1056/NEJMoa2309676
- Locatelli, F., Lang, P., Wall, D., Meisel, R., Corbacioglu, S., Li, A. M., de la Fuente, J., Shah, A. J., Carpenter, B., Kwiatkowski, J. L., Mapara, M., Liem, R. I., Cappellini, M. D., Algeri, M., Kattamis, A., Sheth, S., Grupp, S., Handgretinger, R., Kohli, P., Shi, D., … CLIMB THAL-111 Study Group (2024). Exagamglogene Autotemcel for Transfusion-Dependent β-Thalassemia. The New England journal of medicine, 390(18), 1663–1676. https://doi.org/10.1056/NEJMoa2309673
- Locatelli, F., Thompson, A. A., Kwiatkowski, J. L., Porter, J. B., Thrasher, A. J., Hongeng, S., Sauer, M. G., Thuret, I., Lal, A., Algeri, M., Schneiderman, J., Olson, T. S., Carpenter, B., Amrolia, P. J., Anurathapan, U., Schambach, A., Chabannon, C., Schmidt, M., Labik, I., Elliot, H., … Walters, M. C. (2022). Betibeglogene Autotemcel Gene Therapy for Non-β0/β0 Genotype β-Thalassemia. The New England journal of medicine, 386(5), 415–427. https://doi.org/10.1056/NEJMoa2113206
- Galanello, R., & Origa, R. (2010). Beta-thalassemia. Orphanet journal of rare diseases, 5, 11. https://doi.org/10.1186/1750-1172-5-11
- Needs T, Gonzalez-Mosquera LF, Lynch DT. Beta Thalassemia. [Updated 2023 May 1]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2026 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK531481/
- Langer AL. Beta-Thalassemia. 2000 Sep 28 [Updated 2026 Feb 12]. In: Adam MP, Bick S, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1426/
- Kattamis, A., Forni, G. L., Aydinok, Y., & Viprakasit, V. (2020). Changing patterns in the epidemiology of β-thalassemia. European journal of haematology, 105(6), 692–703. https://doi.org/10.1111/ejh.13512
- Kautz, L., Jung, G., Valore, E. V., Rivella, S., Nemeth, E., & Ganz, T. (2014). Identification of erythroferrone as an erythroid regulator of iron metabolism. Nature genetics, 46(7), 678–684. https://doi.org/10.1038/ng.2996
- Anderson, L. J., Holden, S., Davis, B., Prescott, E., Charrier, C. C., Bunce, N. H., Firmin, D. N., Wonke, B., Porter, J., Walker, J. M., & Pennell, D. J. (2001). Cardiovascular T2-star (T2*) magnetic resonance for the early diagnosis of myocardial iron overload. European heart journal, 22(23), 2171–2179. https://doi.org/10.1053/euhj.2001.2822
- Farmakis, D., Porter, J., Taher, A., Domenica Cappellini, M., Angastiniotis, M., & Eleftheriou, A. (2022). 2021 Thalassaemia International Federation Guidelines for the Management of Transfusion-dependent Thalassemia. HemaSphere, 6(8), e732. https://doi.org/10.1097/HS9.0000000000000732
- Kattamis, A., Kwiatkowski, J. L., & Aydinok, Y. (2022). Thalassaemia. Lancet (London, England), 399(10343), 2310–2324. https://doi.org/10.1016/S0140-6736(22)00536-0
- Agios Pharmaceuticals. (2025, December 23). U.S. FDA Approves Agios' AQVESME™ (mitapivat) for the Treatment of Anemia in Adults with Alpha- or Beta-Thalassemia [Press release]. This is the primary regulatory source confirming the late-2025 FDA approval for both transfusion-dependent (TDT) and non-transfusion-dependent (NTDT) forms of the disease.
- Taher, A. T., Al-Samkari, H., Aydinok, Y., Besser, M., Boscoe, A. N., Dahlin, J. L., De Luna, G., Estepp, J. H., Gheuens, S., Gilroy, K. S., Glenthøj, A., Sim Goh, A., Iyer, V., Kattamis, A., Loggetto, S. R., Morris, S., Musallam, K. M., Osman, K., Ricchi, P., Salido-Fiérrez, E., … ENERGIZE investigators (2025). Mitapivat in adults with non-transfusion-dependent α-thalassaemia or β-thalassaemia (ENERGIZE): a phase 3, international, randomised, double-blind, placebo-controlled trial. Lancet (London, England), 406(10498), 33–42. https://doi.org/10.1016/S0140-6736(25)00635-X
- Cappellini, M. D., et al. (2024). ENERGIZE-T: a global, phase 3, double-blind, randomized, placebo-controlled study of mitapivat in adults with transfusion-dependent alpha- or beta-thalassemia. Blood, 144(Supplement 1), 409. https://doi.org/10.1182/blood-2024-200867
- Federation of Obstetric and Gynaecological Societies of India (FOGSI). (2025). Good Clinical Practice Recommendations (GCPR) on Antenatal Screening for Thalassemia and Hemoglobinopathies.
- Shah, F. T., Nicolle, S., Garg, M., Pancham, S., Lieberman, G., Anthony, K., & Mensah, A. K. (2024). Guideline for the management of conception and pregnancy in thalassaemia syndromes: A British Society for Haematology Guideline. British journal of haematology, 204(6), 2194–2209. https://doi.org/10.1111/bjh.19362