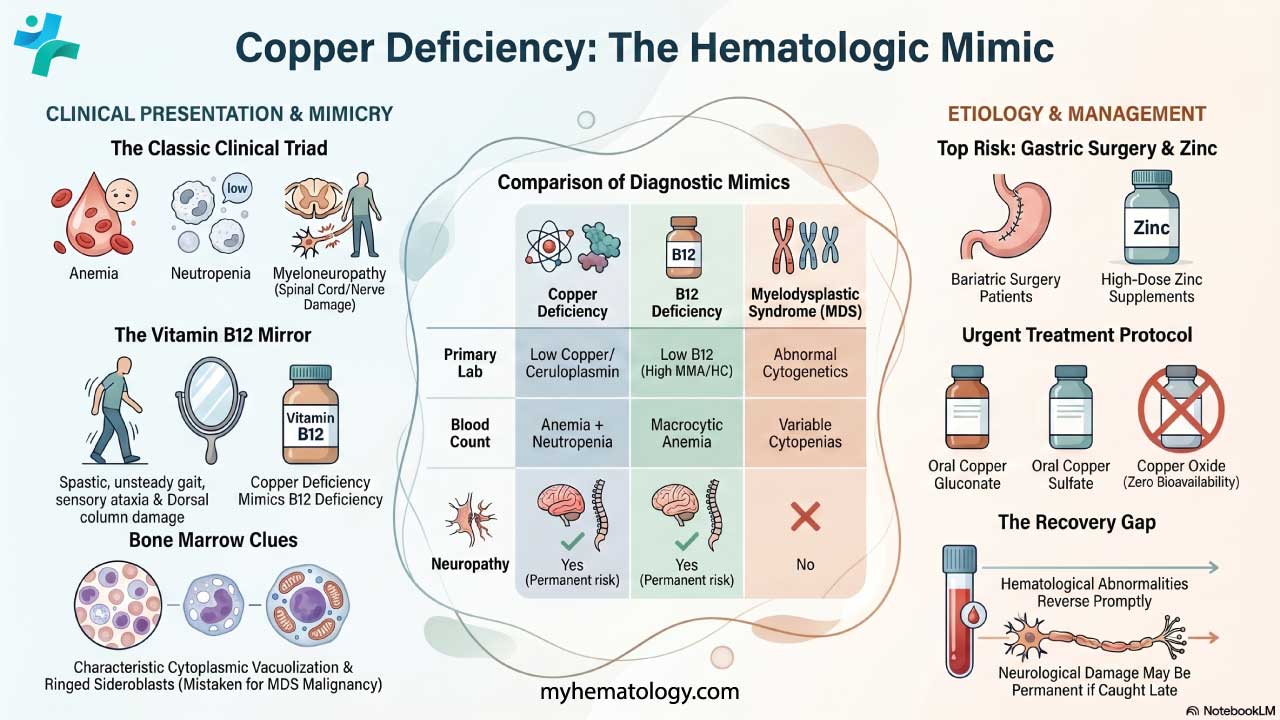
Key Takeaways
Alpha thalassemia is an inherited blood disorder in which one or more of the four alpha-globin genes is missing or faulty, reducing the alpha-globin protein needed to build normal hemoglobin [1].
- Epidemiology ▾: It follows an autosomal recessive inheritance pattern, with the highest carrier rates in Southeast Asia, the Mediterranean, the Middle East, parts of Africa, and the Pacific Islands [2].
- Symptoms of Alpha Thalassemia ▾: Severity depends on how many alpha-globin genes are affected: from silent carriers (one gene) and mild trait (two genes) to hemoglobin H disease (three genes) and Hb Barts hydrops fetalis (all four genes) [1,3].
- Laboratory Investigations ▾: Diagnosis combines a complete blood count, peripheral blood smear, brilliant cresyl blue staining, hemoglobin analysis by HPLC or capillary electrophoresis, and DNA testing to confirm the mutation [3].
- Treatment and Management ▾: Treatment ranges from no intervention in mild forms to folate, transfusions, iron chelation, and splenectomy in HbH disease; hematopoietic stem cell transplantation remains the only established cure for severe alpha thalassemia [3,4]. Modern care for Hb Barts hydrops fetalis offers three counseling options — termination, expectant management, or fetal intervention with intrauterine transfusion and possible postnatal stem cell transplantation [4,5].
*Click ▾ for more information
What is Alpha Thalassemia?
Alpha thalassemia is an inherited form of hemolytic anemia. It happens when the genes that make alpha-globin which is one of the two protein chains in adult hemoglobin are deleted or mutated, so the body produces too little (or no) alpha-globin [1]. Without enough alpha-globin, red blood cells cannot make functional hemoglobin tetramers, and oxygen delivery suffers.
Alpha thalassemia sits within a larger group of conditions called hemoglobinopathies, which affect the structure or production of hemoglobin. The major hemoglobinopathies include sickle cell disease, the thalassemias (alpha and beta), and various structural hemoglobin variants such as hemoglobin E.
Global Prevalence and Significance of Alpha Thalassemia

Alpha thalassemia is one of the most prevalent single-gene blood disorders worldwide, affecting an estimated 270 million carriers, with the highest prevalence in Southeast Asia, the Mediterranean region, and parts of Africa and the Middle East. This widespread prevalence underscores the significant global burden of alpha thalassemia and its impact on individuals and healthcare systems worldwide.
Genetic Basis of Alpha Thalassemia
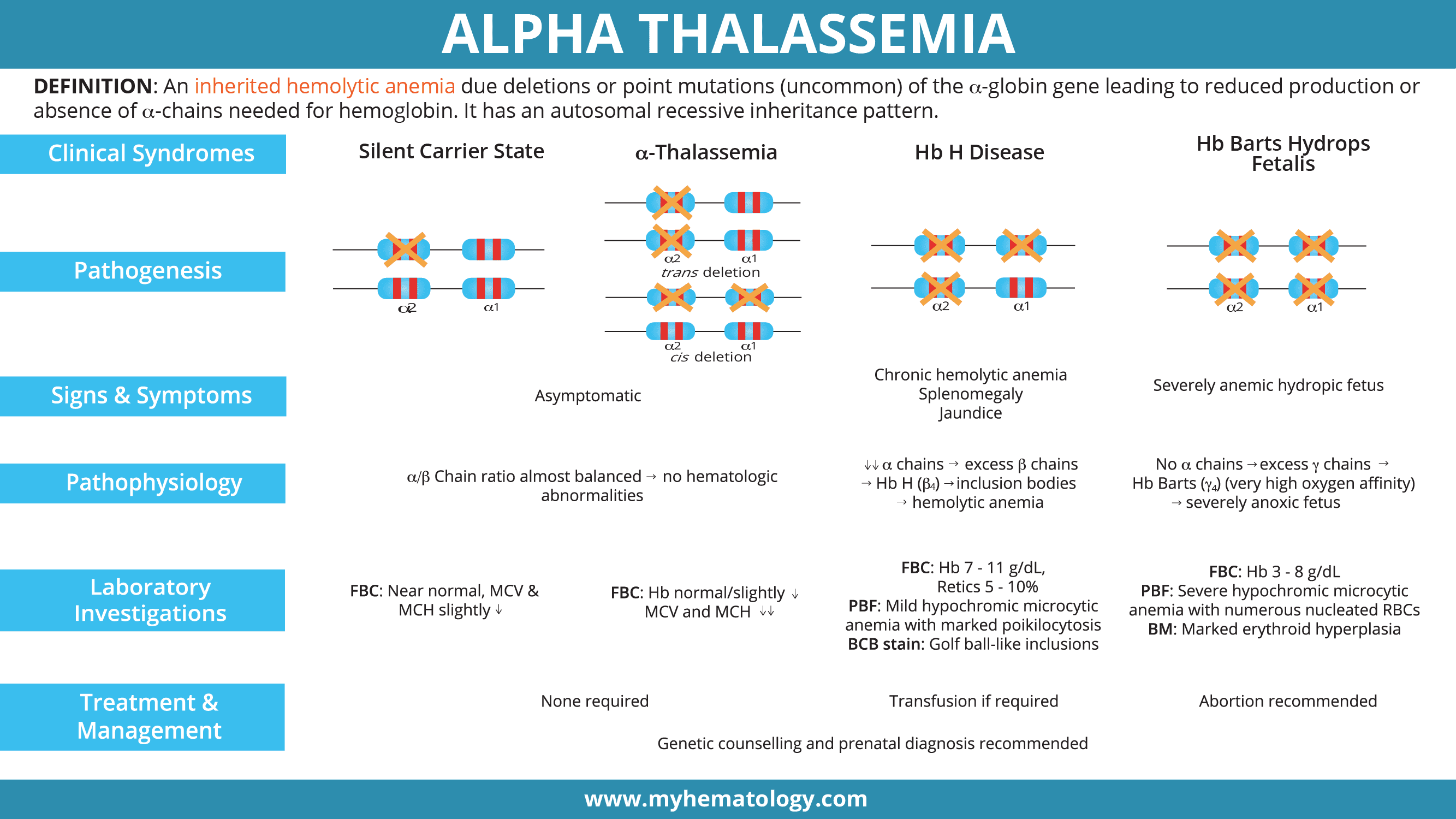
Each person normally inherits four alpha-globin genes: two on each copy of chromosome 16. The two genes on a single chromosome are called HBA1 and HBA2, and the normal genotype is written as αα/αα [1]. Mutations affecting HBA2 tend to cause more severe disease because HBA2 produces the majority of alpha chains.
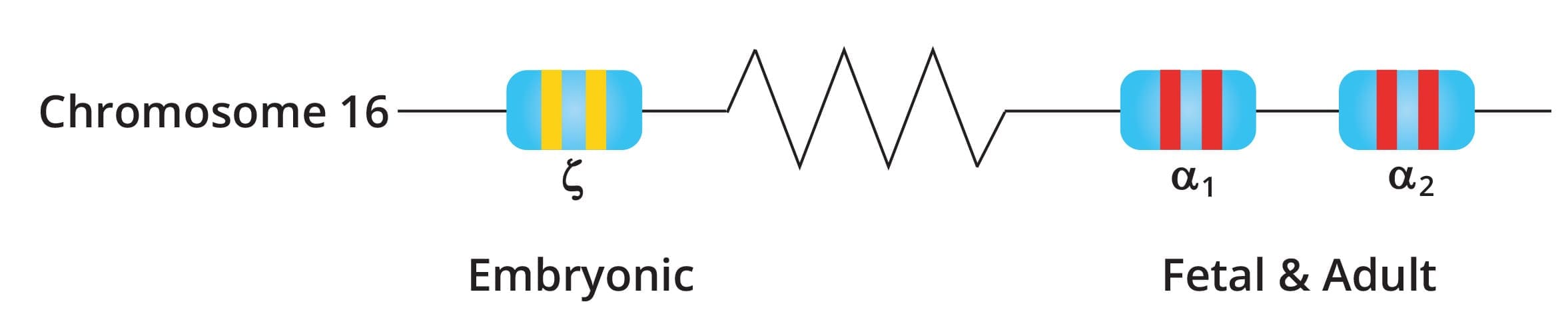
Types of mutations
Alpha thalassemia mutations fall into three main categories [1,2]:
- α⁰-thalassemia (--/αα): both alpha genes on the same chromosome are deleted.
- α⁺-thalassemia (-α/αα): a single alpha gene is deleted.
- Non-deletional α-thalassemia (αᵀα/αα): a point mutation produces an unstable variant such as Hemoglobin Constant Spring or Hemoglobin Adana. Non-deletional forms tend to be more clinically severe because the abnormal protein damages the red cell membrane and triggers oxidative stress [2].
How alpha thalassemia is inherited
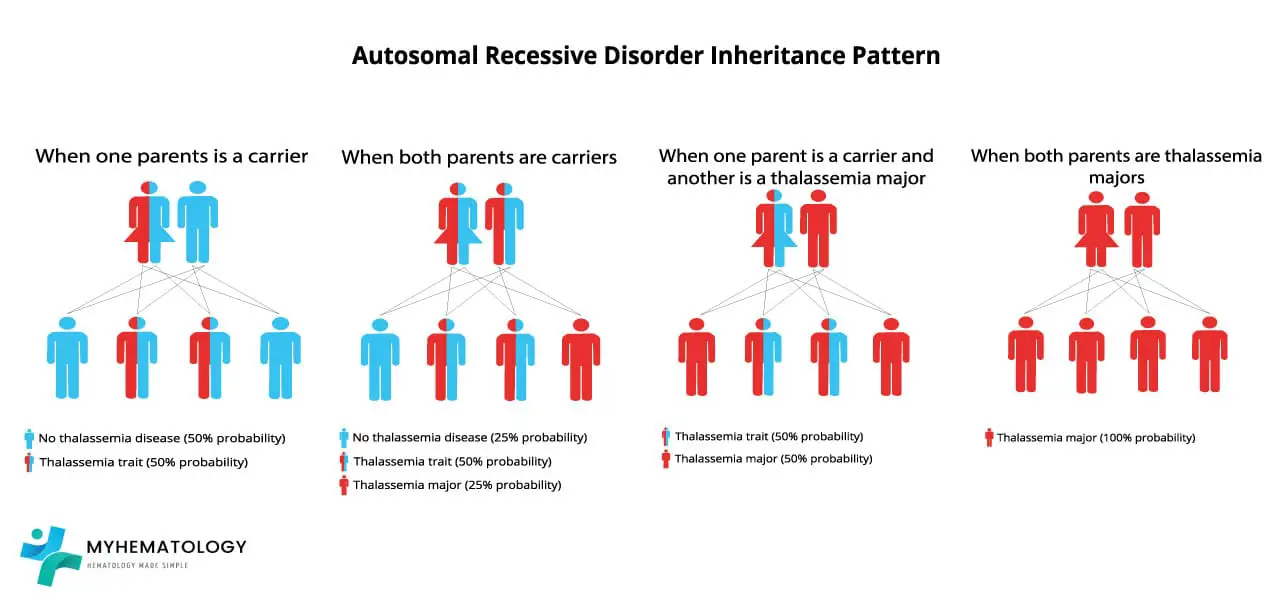
Alpha thalassemia is autosomal recessive. Both parents typically carry at least one affected chromosome, and the child's phenotype depends on the combination they inherit. The cleanest way to predict outcomes is to think of each parent contributing one chromosome (with two alpha genes on it) to the child.
For example, if both parents carry an α⁰ deletion in cis (--/αα), each pregnancy has a 25% chance of a normal child (αα/αα), a 50% chance of a child with alpha thalassemia trait (--/αα), and a 25% chance of Hb Barts hydrops fetalis (--/--). This combination is most common in Southeast Asian populations and is a key reason carrier screening matters in those regions [4].
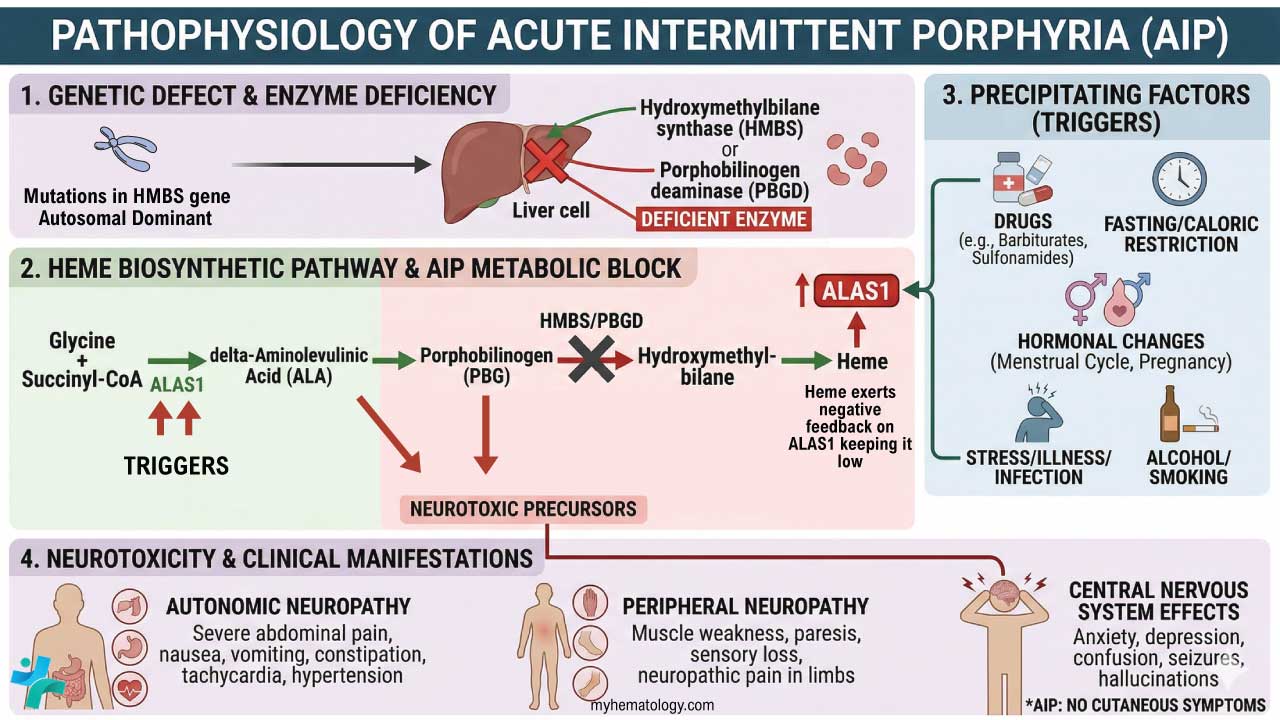
Pathogenesis of Alpha Thalassemia
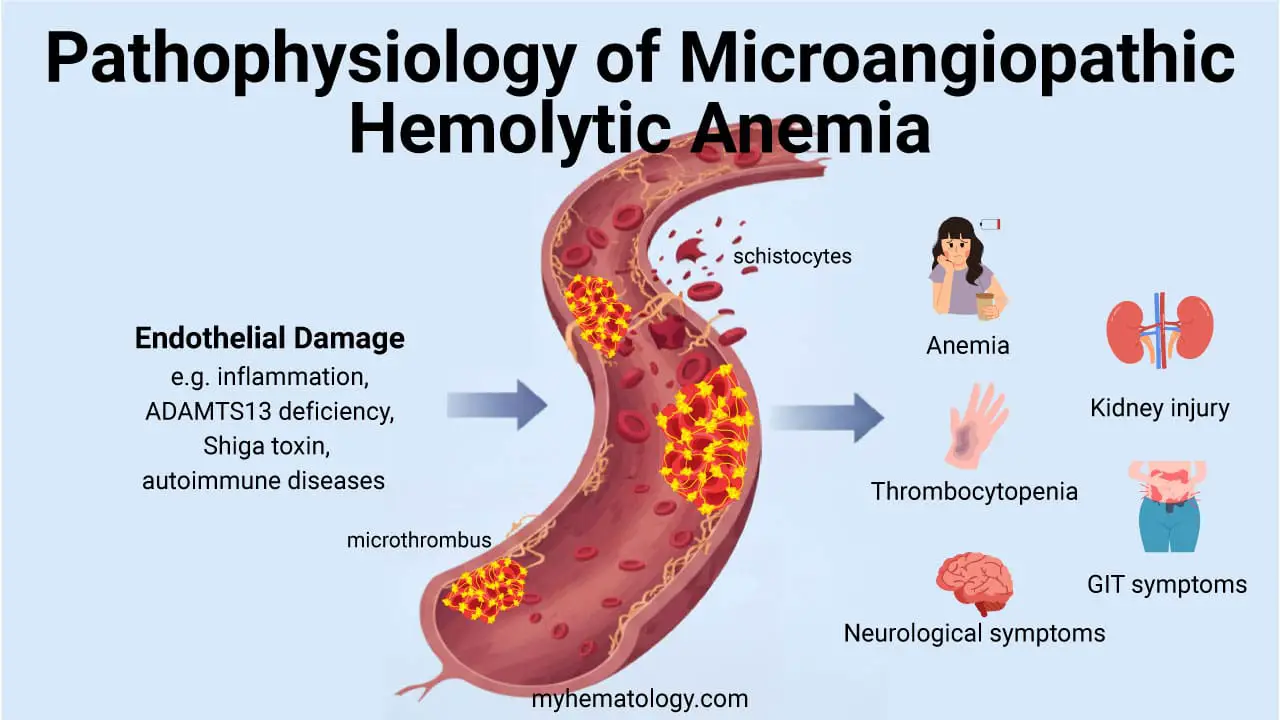
When alpha-globin production drops, two problems occur. First, less normal adult hemoglobin (HbA, α₂β₂) is made, so the blood carries less oxygen. Second, the unmatched globin chains do not simply disappear, instead they form abnormal tetramers that themselves cause harm.
- In adults, excess beta chains form hemoglobin H (β₄). HbH is unstable, precipitates inside aging red blood cells, and triggers their destruction in the spleen.
- In fetuses, excess gamma chains form hemoglobin Barts (γ₄). Hb Barts binds oxygen so tightly it cannot release it to fetal tissues, causing severe tissue hypoxia [1,4].
Because alpha-globin is needed from very early fetal life (unlike beta-globin, which only rises after birth), severe alpha-globin loss has devastating effects in the womb.
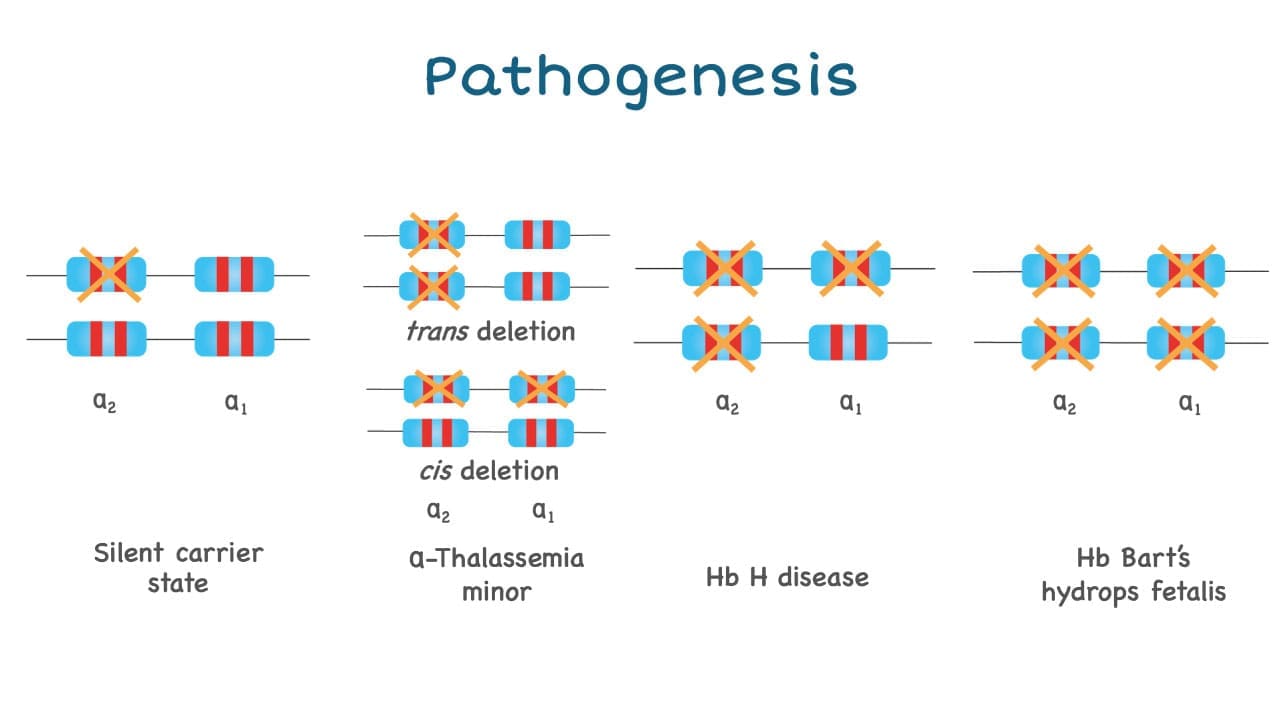
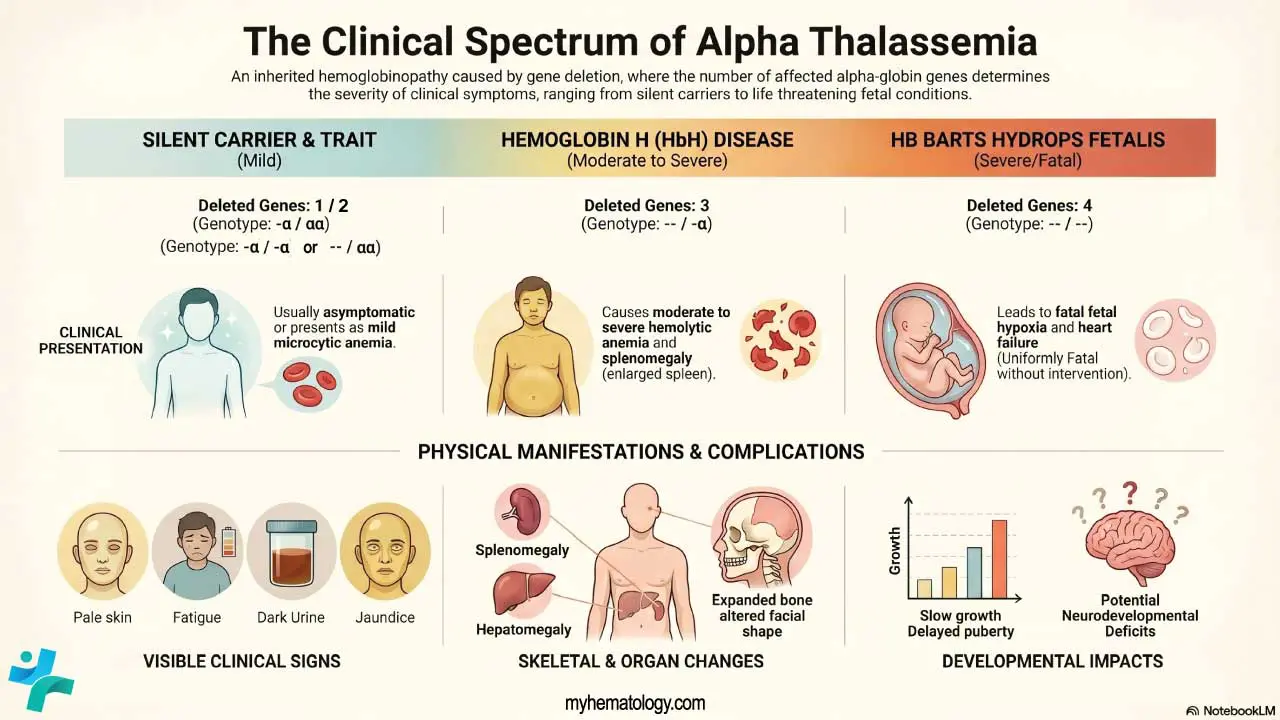
Clinical Picture: Four Phenotypes
The four classic phenotypes correspond to the number of affected alpha-globin genes:
Usually asymptomatic. Red cell indices may show very mild reductions.
Mild microcytic hypochromic anemia, often mistaken for iron deficiency. Mostly asymptomatic [3].
Moderate to severe hemolytic anemia, splenomegaly, jaundice, fatigue, leg ulcers. Worsens with infection, fever, or oxidant drugs [3].
Severe fetal anemia, hydrops, heart failure, urogenital abnormalities. Without fetal intervention, almost uniformly fatal [4].
In moderate to severe forms, common signs include fatigue, pale or jaundiced skin, an enlarged spleen and liver, slow growth and delayed puberty in children, bone marrow expansion (which can change the shape of the face and skull), dark urine, poor appetite, and shortness of breath on exertion.
Diagnosing Alpha Thalassemia
A useful way to learn the lab evaluation is to follow it as a clinician would, from screening to confirmation.
Step 1: Complete blood count (CBC) and red cell indices
The first clue is a microcytic hypochromic anemia — small, pale red cells. In alpha thalassemia trait, the MCV is typically 65–76 fL and the MCH around 22 pg [3].
A high-yield teaching point is the Mentzer index (MCV ÷ RBC count). A value below 13 suggests thalassemia trait; above 13 points to iron deficiency. This is one of the simplest ways to avoid misdiagnosing a thalassemia carrier as iron-deficient and inappropriately starting iron supplements. However, a patient can have both thalassemia trait and iron deficiency simultaneously. Always confirm iron status with a serum ferritin test before relying solely on red cell indices [9].
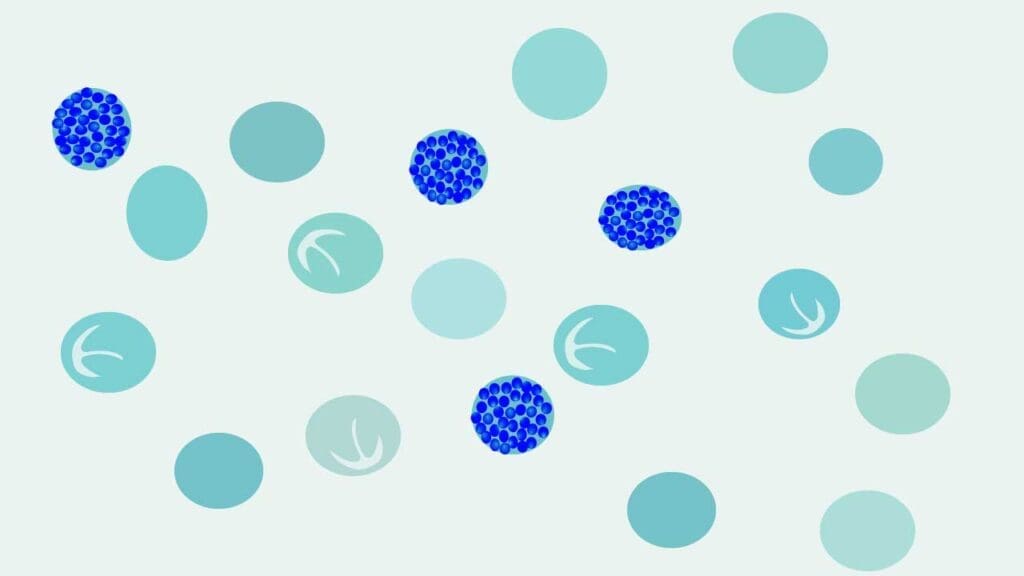
Step 2: Peripheral blood smear
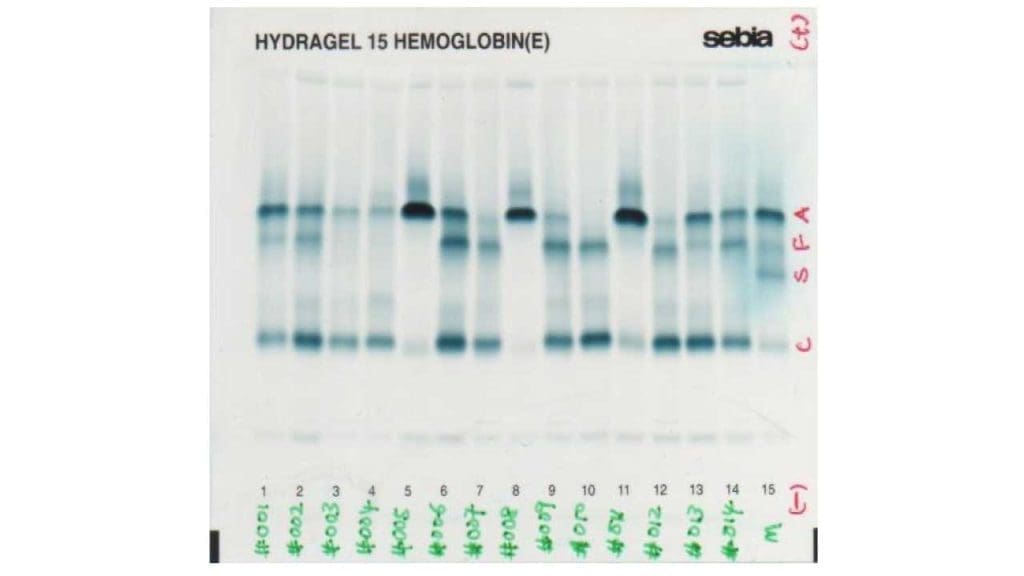
A blood smear shows microcytic hypochromic red cells, target cells, and varying degrees of poikilocytosis (irregular shapes). In HbH disease, smears stained with brilliant cresyl blue (BCB) reveal characteristic golf-ball–like inclusions inside red cells [3]. These are precipitated HbH tetramers.
Step 3: Hemoglobin analysis
HPLC and capillary electrophoresis are the modern workhorses. Both separate hemoglobin variants by charge or size and quantify each subtype. Hemoglobin H and Hemoglobin Barts elute very early in the run, appearing as fast-moving peaks.
Older gel-based hemoglobin electrophoresis (alkaline and acid) is still used in some settings, but automated HPLC and capillary electrophoresis have largely replaced it because they are faster, more reproducible, and easier to interpret.
Step 4: DNA testing
A definitive diagnosis requires identifying the underlying mutation. GAP-PCR detects common deletional mutations, while multiplex ligation-dependent probe amplification (MLPA) or direct sequencing is used for rare deletions and non-deletional mutations like Constant Spring [2].
Increasingly, Next-Generation Sequencing (NGS) and long-read sequencing technologies are becoming the gold standards in advanced diagnostic centers due to their superior ability to detect complex structural variants and rare non-deletional mutations that traditional PCR misses [10].
Lab findings by phenotype
- Silent carrier: near-normal CBC; often missed without DNA testing.
- Alpha thalassemia trait: mild anemia, low MCV and MCH, microcytic hypochromic smear.
- HbH disease: Hb 7–11 g/dL, reticulocytes 5–10%, marked poikilocytosis, target cells, HbH inclusions on BCB stain, HbH peak on HPLC.
- Hb Barts hydrops fetalis: Hb 3–8 g/dL, severe poikilocytosis, nucleated red cells, marked erythroid hyperplasia in bone marrow, dominant Hb Barts on hemoglobin analysis [4].
Treatment and Management
Management depends entirely on phenotype.
Mild forms
Silent carriers and people with alpha thalassemia trait usually need no treatment. Folic acid supplementation may be offered to support red cell production. Crucially, iron supplements should not be given unless iron deficiency is confirmed, because alpha thalassemia patients are at risk of iron overload.
Hemoglobin H disease
Standard care includes [3]:
- Folic acid supplementation to support erythropoiesis.
- Transfusions as needed, particularly during infections, fever, pregnancy, or surgery. Some patients become transfusion-dependent.
- Iron chelation therapy with deferasirox, deferiprone (oral), or deferoxamine (intravenous) for transfusion-related iron overload.
- Splenectomy in selected cases of marked splenomegaly or rising transfusion needs, balanced against the lifelong infection risk.
- Avoidance of oxidant drugs (such as sulfonamides and certain antimalarials), which can trigger hemolytic crises.
Disease-modifying therapies have rapidly reshaped the treatment picture. On December 23, 2025, the FDA approved mitapivat (AQVESME), an oral pyruvate kinase activator, for adults with both non-transfusion-dependent and transfusion-dependent alpha- and beta-thalassemia [11]. This landmark approval was based on the Phase 3 ENERGIZE and ENERGIZE-T trials, which demonstrated significant improvements in hemoglobin levels and reductions in transfusion burden [12]. Furthermore, luspatercept, a TGF-β ligand trap FDA-approved for beta-thalassemia, reported positive Phase 2 top-line results for alpha-thalassemia in early 2026, though its use in alpha-thalassemia remains off-label pending final regulatory review [13]. Recent CRISPR gene therapies are currently approved only for beta-thalassemia and sickle cell disease, not alpha-thalassemia.
Hb Barts hydrops fetalis (alpha thalassemia major)
This was once considered uniformly fatal. That has changed. Modern management offers families three options after prenatal diagnosis [4,5]:
- Termination of pregnancy, supported by counseling.
- Expectant management with full counseling about likely fetal demise and severe maternal risks. Specifically, practitioners must warn families about Mirror Syndrome (maternal hydrops), a life-threatening condition where the mother's body begins to "mirror" the sick fetus, dramatically elevating the risk of severe pre-eclampsia and postpartum hemorrhage [14].
- Fetal intervention with serial intrauterine transfusions to correct fetal anemia and prevent hydrops, followed by lifelong postnatal transfusions or hematopoietic stem cell transplantation from a matched donor as the only established cure. An FDA-approved phase 1 trial at UCSF is also testing in utero maternal stem cell transplantation [5].
Survivors of intrauterine transfusion can live well into childhood and beyond, but families should be counseled that long-term morbidity including growth retardation, developmental delay, neurological deficits, and a high rate of hypospadias in male infants remains a real risk [4].
Prenatal Screening: Why It Matters
In high-prevalence regions, carrier screening prevents tragic outcomes. The pathway is straightforward in principle: a low MCV on a routine CBC triggers hemoglobin analysis. If suggestive of alpha thalassemia, DNA testing confirms the genotype. The partner is then tested. When both partners carry α⁰-thalassemia in cis, prenatal diagnosis by chorionic villus sampling or amniocentesis is offered, ideally in the first trimester, so that families have time to consider all management options [4,8].
Alpha Thalassemia Trait vs Iron Deficiency Anemia
Both produce small, pale red cells, and the two are routinely confused in primary care. Two findings help separate them:
- Mentzer index < 13 favors thalassemia; > 13 favors iron deficiency.
- Iron studies: ferritin and transferrin saturation are normal or elevated in thalassemia trait, but low in iron deficiency.
Definitive distinction requires hemoglobin analysis and, where indicated, DNA testing. Getting this right matters because giving iron to a thalassemia carrier is at best useless and at worst harmful.
Frequently Asked Questions (FAQs)
What is alpha thalassemia in simple terms?
Alpha thalassemia is an inherited blood disorder in which the body makes too little (or none) of the alpha-globin protein needed to build hemoglobin, the molecule that carries oxygen in red blood cells. The severity depends on how many of the four alpha-globin genes are missing or faulty, ranging from no symptoms (one gene affected) to a life-threatening fetal condition (all four affected).
How is alpha thalassemia diagnosed?
Diagnosis usually starts with a complete blood count showing small, pale red blood cells (microcytic hypochromic anemia). A peripheral blood smear and brilliant cresyl blue stain can reveal hemoglobin H inclusion bodies. Hemoglobin analysis by HPLC or capillary electrophoresis detects abnormal hemoglobins like HbH or Hb Barts. Final confirmation comes from DNA testing — GAP-PCR for deletional mutations and sequencing or MLPA for non-deletional ones.
Should people with alpha thalassemia take iron supplements?
Generally no. The anemia in alpha thalassemia is caused by faulty hemoglobin production, not by iron deficiency. Taking iron supplements without proven deficiency can lead to iron overload, which damages the heart, liver, and endocrine glands. Iron should only be taken if blood tests confirm true iron deficiency, and only under medical supervision.
Is alpha thalassemia curable?
For most carriers and people with alpha thalassemia trait, no cure is needed because they have no symptoms. For HbH disease, treatment focuses on transfusions, folate, and chelation as needed. The only established curative treatment for severe alpha thalassemia is allogeneic hematopoietic stem cell transplantation from a matched donor. Gene therapy approaches are under active study but are not yet approved for alpha thalassemia.
Can a baby with alpha thalassemia major (Hb Barts hydrops fetalis) survive?
Without intervention, alpha thalassemia major is almost always fatal before or shortly after birth. With early prenatal diagnosis and serial intrauterine blood transfusions, fetal survival is now possible, and some children have gone on to receive curative stem cell transplants after birth [4,5]. Survivors often have neurodevelopmental and physical complications, so families need detailed counseling about all three options: continuing pregnancy with fetal intervention, expectant management, or termination.
How do you tell alpha thalassemia trait apart from iron deficiency anemia?
Both cause small, pale red blood cells, so a CBC alone cannot reliably tell them apart. Two clues help. First, the Mentzer index (MCV ÷ RBC count): a value below 13 favors thalassemia trait, above 13 favors iron deficiency. Second, iron studies (ferritin, transferrin saturation) are normal or high in thalassemia trait but low in iron deficiency. Definitive distinction requires hemoglobin analysis and, when needed, DNA testing.
Glossary of Related Medical Terms
- Allele — one of two copies of a gene a person inherits, one from each parent.
- Autosomal recessive inheritance — a pattern where a person needs two faulty copies of a gene (one from each parent) to show the disease.
- Cis vs trans deletions — cis means both deleted alpha genes sit on the same chromosome; trans means one deletion sits on each of the two chromosomes. Cis deletions in both parents create the highest risk for hydrops fetalis.
- Erythropoiesis — the body's process of making red blood cells, mainly in the bone marrow.
- Hemoglobin (Hb) — the iron-containing protein inside red blood cells that carries oxygen.
- Hemoglobin Bart's (Hb Barts) — an abnormal hemoglobin made of four gamma chains (γ₄). It binds oxygen so tightly it cannot release it to tissues.
- Hemoglobin H (HbH) — an abnormal hemoglobin made of four beta chains (β₄). It is unstable and damages red blood cells from the inside.
- HPLC (high-performance liquid chromatography) — a lab technique that separates and measures different types of hemoglobin.
- Hydrops fetalis — severe fluid buildup in a fetus, often a sign of life-threatening anemia in alpha thalassemia major.
- Iron chelation therapy — medication that binds excess iron in the body so it can be excreted, used in patients receiving regular blood transfusions.
- Mentzer index — MCV ÷ red blood cell count; a value below 13 suggests thalassemia trait, above 13 suggests iron deficiency.
- Microcytic hypochromic anemia — anemia where red blood cells are smaller (microcytic) and paler (hypochromic) than normal.
- Poikilocytosis — variation in red blood cell shape on a blood smear.
- Splenectomy — surgical removal of the spleen.
- Target cells — red blood cells that look like archery targets on a smear, common in thalassemia.
Disclaimer: This article is intended for educational and informational purposes only. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Higgs D. R. (2013). The molecular basis of α-thalassemia. Cold Spring Harbor perspectives in medicine, 3(1), a011718. https://doi.org/10.1101/cshperspect.a011718
- Kalle Kwaifa, I., Lai, M. I., & Md Noor, S. (2020). Non-deletional alpha thalassaemia: a review. Orphanet journal of rare diseases, 15(1), 166. https://doi.org/10.1186/s13023-020-01429-1
- Songdej, D., & Fucharoen, S. (2022). Alpha-Thalassemia: Diversity of Clinical Phenotypes and Update on the Treatment. Thalassemia Reports, 12(4), 157-172. https://doi.org/10.3390/thalassrep12040020
- Vichinsky E. P. (2009). Alpha thalassemia major--new mutations, intrauterine management, and outcomes. Hematology. American Society of Hematology. Education Program, 35–41. https://doi.org/10.1182/asheducation-2009.1.35
- Winger, B. A., Ajayi, A., & Vichinsky, E. (2025). Diagnosis and Treatment of Alpha Thalassemia Major. Hemoglobin, 49(1), 3–9. https://doi.org/10.1080/03630269.2024.2432899
- Kuo, K. H. M., Layton, D. M., Lal, A., Al-Samkari, H., Bhatia, J., Kosinski, P. A., Tong, B., Lynch, M., Uhlig, K., & Vichinsky, E. P. (2022). Safety and efficacy of mitapivat, an oral pyruvate kinase activator, in adults with non-transfusion dependent α-thalassaemia or β-thalassaemia: an open-label, multicentre, phase 2 study. Lancet (London, England), 400(10351), 493–501. https://doi.org/10.1016/S0140-6736(22)01337-X
- Cappellini, M. D., Viprakasit, V., Taher, A. T., Georgiev, P., Kuo, K. H. M., Coates, T., Voskaridou, E., Liew, H. K., Pazgal-Kobrowski, I., Forni, G. L., Perrotta, S., Khelif, A., Lal, A., Kattamis, A., Vlachaki, E., Origa, R., Aydinok, Y., Bejaoui, M., Ho, P. J., Chew, L. P., … BELIEVE Investigators (2020). A Phase 3 Trial of Luspatercept in Patients with Transfusion-Dependent β-Thalassemia. The New England journal of medicine, 382(13), 1219–1231. https://doi.org/10.1056/NEJMoa1910182
- Amid A, Lal A, Coates TD, et al., editors. Guidelines for the Management of α-Thalassaemia [Internet]. Nicosia (Cyprus): Thalassaemia International Federation; 2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK602223/
- Vehapoglu, A., Ozgurhan, G., Demir, A. D., Uzuner, S., Nursoy, M. A., Turkmen, S., & Kacan, A. (2014). Hematological indices for differential diagnosis of Beta thalassemia trait and iron deficiency anemia. Anemia, 2014, 576738. https://doi.org/10.1155/2014/576738
- Shang, X., Peng, Z., Ye, Y., Asan, Zhang, X., Chen, Y., Zhu, B., Cai, W., Chen, S., Cai, R., Guo, X., Zhang, C., Zhou, Y., Huang, S., Liu, Y., Chen, B., Yan, S., Chen, Y., Ding, H., Yin, X., … Xu, X. (2017). Rapid Targeted Next-Generation Sequencing Platform for Molecular Screening and Clinical Genotyping in Subjects with Hemoglobinopathies. EBioMedicine, 23, 150–159. https://doi.org/10.1016/j.ebiom.2017.08.015
- U.S. Food and Drug Administration. (2025, December 23). FDA approves first oral treatment for anemia in thalassemia, an inherited blood disorder. FDA.
- Taher, A. T., Al-Samkari, H., Aydinok, Y., Besser, M., Boscoe, A. N., Dahlin, J. L., De Luna, G., Estepp, J. H., Gheuens, S., Gilroy, K. S., Glenthøj, A., Sim Goh, A., Iyer, V., Kattamis, A., Loggetto, S. R., Morris, S., Musallam, K. M., Osman, K., Ricchi, P., Salido-Fiérrez, E., … ENERGIZE investigators (2025). Mitapivat in adults with non-transfusion-dependent α-thalassaemia or β-thalassaemia (ENERGIZE): a phase 3, international, randomised, double-blind, placebo-controlled trial. Lancet (London, England), 406(10498), 33–42. https://doi.org/10.1016/S0140-6736(25)00635-X
- Bristol Myers Squibb. (2026, February 23). Bristol Myers Squibb Announces Positive Top-Line Results from Registrational Phase 2 Study of Luspatercept in Adults with Alpha (α)-Thalassemia [Press release].
- Han, Z., Chen, X., Wang, Q., Zhou, J., Guo, Y., Hou, H., & Zhang, Y. (2021). Clinical characteristics and risk factors of mirror syndrome: a retrospective case-control study. BMC pregnancy and childbirth, 21(1), 660. https://doi.org/10.1186/s12884-021-04143-3