Key Takeaways
Hereditary hemochromatosis is a genetic disorder of iron regulation that causes progressive iron overload in organs and tissues.
- Causes ▾: Most cases involve HFE gene mutations (C282Y/H63D); inheritance is autosomal recessive.
- Symptoms ▾:
- Early symptoms are often non-specific (fatigue, weakness, joint pain).
- Late complications include cirrhosis, cardiomyopathy, diabetes, joint damage, skin bronzing.
- Diagnosis ▾: Fasting transferrin saturation (>45%), serum ferritin, HFE genetic testing.
- Treatment and Management ▾: Therapeutic phlebotomy (first line); chelation therapy (second line).
- Prognosis ▾: Excellent with early diagnosis; cirrhosis significantly worsens outcomes.
*Click ▾ for more information
Introduction
Hemochromatosis is the most common genetic cause of this kind of iron overload. In hereditary hemochromatosis (HH), inherited mutations stop the body from sensing how much iron it already has. So the gut keeps absorbing iron, no matter how full the stores are. Over years and decades, iron builds up in the liver, heart, pancreas, joints, and hormone-producing glands. The consequences can be life-threatening.[4]
Iron is essential for human life. It sits at the core of hemoglobin, the protein inside red blood cells that carries oxygen from the lungs to every tissue. The body also needs iron for energy production, immune defense, and DNA synthesis.
Here is the catch: the body has no active way to get rid of extra iron. The only way to control iron levels is to carefully regulate how much enters the body from food. When that regulation breaks down, iron piles up in the organs. Once there, it generates toxic molecules called reactive oxygen species, which damage cells permanently.[3]
Hereditary hemochromatosis is common, treatable, and often missed. Caught early and managed consistently, most patients live a normal life span. Caught late, the damage to organs may already be permanent.
Prevalence
Hemochromatosis is most common in people of Northern European, especially Celtic, ancestry. About 1 in 300 to 500 non-Hispanic white people in Europe, North America, and Australia carry two copies of the most common causative mutation.[1] Rates are much lower in Hispanic, East Asian, South Asian, Pacific Islander, and sub-Saharan African populations.
There is an important nuance though. Not everyone who inherits the high-risk genotype actually develops iron overload. This is called incomplete penetrance. Studies suggest roughly 25 to 50 percent of C282Y homozygotes (people who inherit two copies of the C282Y mutation) develop biochemical signs of iron overload. A smaller fraction go on to organ damage.[6] Several factors shape who progresses: sex (women lose iron through menstruation, so they accumulate more slowly), alcohol intake, diet, and any other liver disease they may have.
Pathophysiology: The Hepcidin–Ferroportin Axis
To understand hemochromatosis, it helps to know how the body normally controls iron. The master regulator is a small hormone made by the liver called hepcidin.[5] Hepcidin works by binding to a protein called ferroportin, the only known protein that exports iron out of cells. Ferroportin sits on the surface of gut lining cells and macrophages.
When iron stores are full, the liver releases more hepcidin. Hepcidin tags ferroportin for destruction, which blocks gut cells from sending iron into the bloodstream. Absorption drops. When iron is low or red blood cell production is high, hepcidin levels fall. Ferroportin is left intact, and the gut absorbs more iron.
In hemochromatosis, mutations in HFE (and in other genes for non-HFE forms) break the signaling pathway that tells the liver to make hepcidin. The liver does not make enough hepcidin for the amount of iron in the body. Ferroportin is not properly suppressed. Iron keeps flowing in from food, faster than the body needs. Over time, iron overload sets in.[9]
Think of hepcidin as the body's iron thermostat. In hemochromatosis, the thermostat is broken. The body cannot tell that iron stores are already full, so absorption keeps going. This is exactly why phlebotomy (removing iron-rich blood) works as treatment, and why diet alone is not enough.
Causes
The HFE Gene (Type 1 — Most Common)
The HFE gene sits on chromosome 6p21.3. It codes for a protein that is part of the sensing system in liver and gut cells. This system tells the liver how much iron is in the blood, so it can adjust hepcidin accordingly. When the HFE protein does not work properly, the system fails. Hepcidin stays inappropriately low.[4]
C282Y Mutation
The C282Y mutation swaps one amino acid in the HFE protein at position 282: cysteine (C) for tyrosine (Y). That single change stops the HFE protein from binding properly to a partner called beta-2 microglobulin (β2M). Without that partnership, HFE cannot reach the cell surface, and the hepcidin signaling pathway breaks down. Iron absorption runs unchecked. C282Y homozygosity, meaning two copies of this mutation, accounts for roughly 80 to 85 percent of clinical hemochromatosis cases in Northern European populations.[3]
H63D Mutation
The H63D mutation swaps histidine (H) at position 63 for aspartic acid (D). Its effect is milder. The protein still partly binds β2M and interacts with TFR1, so iron regulation is less disrupted. People with two copies of H63D rarely develop significant iron overload. Compound heterozygotes (one C282Y allele and one H63D allele) sometimes develop mild to moderate overload and need monitoring. Their risk of organ damage is still lower than C282Y homozygotes.[10]
Inheritance Pattern
Type 1 hemochromatosis follows an autosomal recessive pattern. Both copies of HFE need a causative mutation for the disease to show. If both parents carry one copy of C282Y (heterozygous carriers), each child has a 25 percent chance of being affected (homozygous), a 50 percent chance of being a carrier, and a 25 percent chance of inheriting neither mutation. Carriers themselves rarely develop iron overload, but they can pass the mutation on.[3,4]
Classification of Hemochromatosis Types
HFE mutations (Type 1) cause most cases. Several rarer hereditary forms involve different genes. The table below summarizes the main types.[3,4]
Table 1. Classification of hereditary hemochromatosis by gene and clinical features. Adapted from EASL (2022)[4] and Brissot et al. (2018)[3].
| Type | Gene | Protein affected | Age of onset | Key features |
| 1 | HFE | Iron-sensing complex | Adult (>40) | Most common; variable penetrance |
| 2A | HJV | Hemojuvelin | <30 years | Juvenile form; severe cardiomyopathy & hypogonadism |
| 2B | HAMP | Hepcidin | <30 years | Juvenile form; rare |
| 3 | TFR2 | Transferrin receptor 2 | Adult | Moderate severity; rare |
| 4 | SLC40A1 | Ferroportin | Adult | Autosomal dominant; may present as macrophage overload |
Hereditary Hemochromatosis Symptoms & Disease Staging
Clinicians use a recognized four-stage framework to map symptoms onto disease progression. This staging helps decide how urgent investigation and treatment should be.[4]
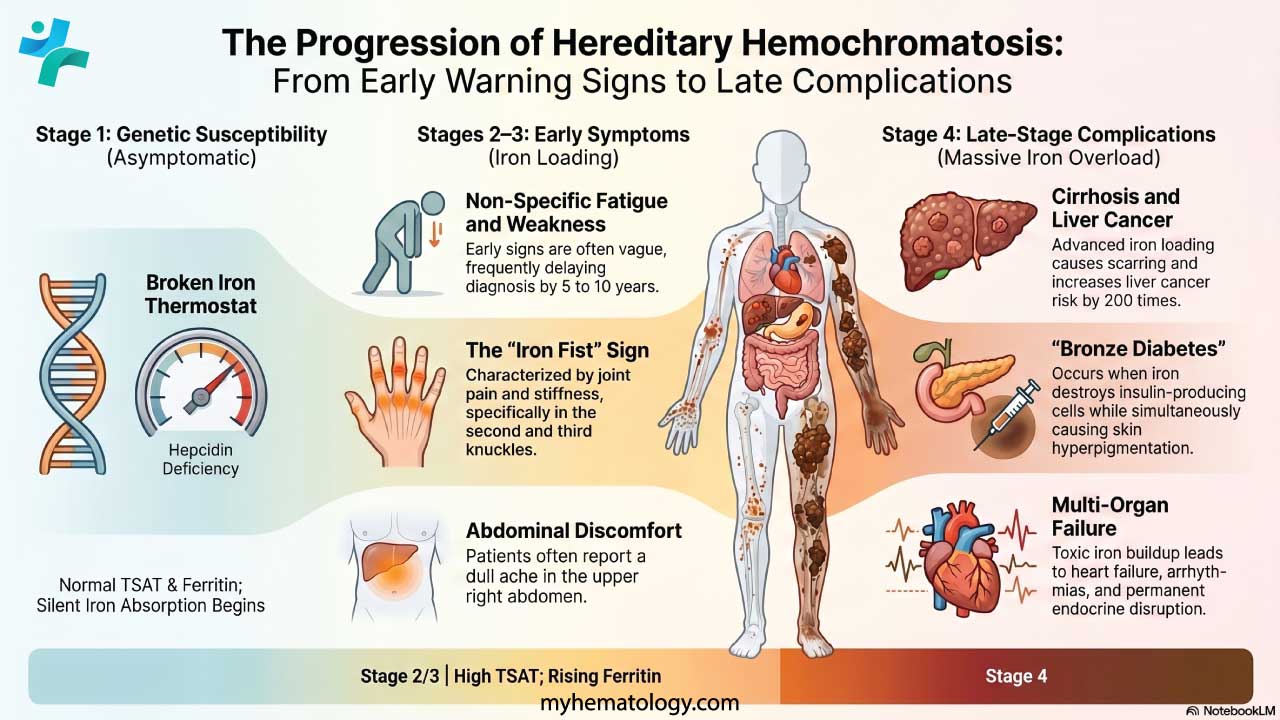
Disease Stages
Table 2. Four-stage progression of hereditary hemochromatosis. Adapted from EASL (2022)[4].
| Stage | Biochemistry | Symptoms | Key action |
| 1 | Normal TSAT & ferritin | None — genetic susceptibility only | Genetic testing if family history |
| 2 | Elevated TSAT; rising ferritin | Usually none; fatigue possible | Confirm with iron studies; begin monitoring |
| 3 | High TSAT & ferritin; early hepatic iron | Fatigue, joint pain, abdominal pain | Start therapeutic phlebotomy |
| 4 | Iron overload in multiple organs | Cirrhosis, cardiomyopathy, diabetes, skin bronzing | Urgent treatment; specialist referral; HCC surveillance |
Early Symptoms (Stages 1–2)
Early symptoms are vague. They look like a dozen other common problems. That is exactly why diagnosis is often delayed by 5 to 10 years from when symptoms first appear.[7] Common complaints include:
- Persistent fatigue and reduced stamina.
- Generalized weakness.
- Joint pain and stiffness, especially in the second and third knuckles of the hand. This pattern is sometimes called the "iron fist" sign.
- A dull ache in the upper right abdomen.
- Unexplained weight loss.
- Cognitive symptoms, often described as "brain fog."
Later Complications (Stages 3–4)
As iron continues to deposit in organs, the picture becomes more specific and more serious:
- Liver: Fat buildup, scarring, and eventually cirrhosis. This brings jaundice, fluid in the abdomen, and high pressure in the liver's blood vessels. Once cirrhosis is established, the risk of liver cancer (hepatocellular carcinoma, or HCC) rises sharply, around 200 times the general population.[2]
- Heart: Iron in heart muscle cells causes the heart to either stiffen or dilate. This leads to abnormal rhythms and heart failure. In untreated juvenile hemochromatosis, heart failure is the leading cause of death.[3]
- Pancreas: Insulin-producing beta cells are destroyed, and diabetes develops. When this happens together with skin pigmentation, it is sometimes called "bronze diabetes."[4]
- Joints: Arthropathy (joint disease) progresses in the hands, hips, and knees. It can be painful and disabling. Unlike many other complications, joint damage often does not improve once iron is removed.[7]
- Skin: Iron stimulates extra melanin, giving the skin a bronze or grayish-brown tone. The change is most visible in sun-exposed areas.[10]
- Endocrine system: Iron disrupts the hypothalamic-pituitary axis. The result is hypogonadotropic hypogonadism, meaning low sex hormone production. This causes reduced libido, erectile dysfunction in men, irregular periods in women, and fertility problems.[4,7]
Why Do Women Present Later Than Men?
Women of reproductive age have a built-in protection: menstruation. Regular blood loss means regular iron loss, which slows down accumulation. Because of this, premenopausal women typically develop symptomatic hemochromatosis 10 to 20 years later than men. After menopause, or in women who do not menstruate (for example, due to premature ovarian insufficiency), iron accumulates faster and symptoms can appear earlier than expected.[1,6]
Laboratory Investigations
Initial Screening
Diagnosis starts with a focused iron profile, drawn in the morning after fasting. Fasting reduces the impact of recent meals on the results.[4]
- Transferrin saturation (TSAT): This is the most sensitive early marker. A fasting TSAT above 45 percent (without another clear cause) is the trigger to investigate further. TSAT is the ratio of serum iron to total iron-binding capacity. It rises early in hemochromatosis, before ferritin climbs.[4,6]
- Serum ferritin: This reflects total body iron stores. A high ferritin (>300 µg/L in men and post-menopausal women; >200 µg/L in pre-menopausal women) along with high TSAT raises the suspicion of hemochromatosis. One important caveat: ferritin is also an acute-phase reactant. It can rise in obesity, metabolic syndrome, alcohol use, fatty liver disease, and inflammation. Always interpret it in clinical context.[4,6]
Confirmatory Tests
- Genetic testing: Testing for C282Y and H63D in the HFE gene is the standard confirmatory test. C282Y homozygosity, combined with elevated TSAT and ferritin, confirms most cases without needing a liver biopsy.[10]
- Liver function tests (LFTs): Raised AST and ALT point to liver inflammation. Importantly, normal LFTs do not rule out iron buildup in the liver, even when ferritin is high.[7]
- Complete blood count (CBC): Usually normal. Anemia is not typical of uncomplicated hemochromatosis. If a patient is anemic, look for another cause or a coexisting condition.[10]
Advanced Staging Investigations
- MRI of the liver (T2*/R2* sequences): A validated, non-invasive way to measure how much iron is in the liver. It is increasingly used to stage iron overload and track treatment response without a biopsy.[4]
- Liver biopsy: Now reserved for specific situations. Use it when (1) the diagnosis is unclear and HFE homozygosity is absent, (2) ferritin is very high (above 1,000 µg/L) and cirrhosis must be ruled out, or (3) you need to stage fibrosis to guide prognosis or management.[4,6]

Interpreting Elevated Ferritin
Hyperferritinemia (raised ferritin) is common, and most causes are not iron overload. Before pinning a high ferritin on hemochromatosis, consider metabolic syndrome or obesity, alcohol use disorder, non-alcoholic fatty liver disease (NAFLD), inflammatory conditions like rheumatoid arthritis or active infection, and blood cancers. Always pair ferritin with transferrin saturation and the full clinical picture.[4,6]
Treatment and Management
Therapeutic Phlebotomy (Venesection)
Phlebotomy is the cornerstone of treatment for anyone with clinical hemochromatosis and confirmed iron overload. Removing 450 to 500 mL of blood takes out about 200 to 250 mg of iron.[7] Treatment runs in two phases:
- Induction phase: Blood is removed weekly, sometimes twice weekly, until iron stores drop. The targets are serum ferritin below 50 µg/L and TSAT below 30 percent. Depending on how much iron has built up, this phase lasts anywhere from 6 months to 2 years.[4,7]
- Maintenance phase: Once iron levels are normal, phlebotomy drops to three or four sessions a year to stop iron from building back up. Iron studies continue every three to six months for life.[4]
In some countries, patients can give blood therapeutically through the national blood service. This has clear benefits: it can boost quality of life, and it lowers the cost of treatment to the health system.[7]
Chelation Therapy
Chelation is used when phlebotomy is not possible or not tolerated. Examples include severe anemia, heart failure, or poor venous access. Chelating agents grab onto iron in the blood so it can leave through urine or stool. The agents include desferrioxamine (given by injection or infusion), deferasirox (oral, the first-line oral option), and deferiprone (oral). Side effects vary: gastrointestinal upset is common, deferasirox can affect the kidneys, and deferiprone can cause agranulocytosis (a dangerous drop in white blood cells). Chelation is much less efficient at removing iron than phlebotomy, so it is not recommended as first-line treatment for HFE-related hemochromatosis.[4]
Dietary Management
Diet plays a supporting role. Phlebotomy removes iron far more effectively than any food restriction. Extreme avoidance of dietary iron is neither necessary nor realistic to keep up. The evidence-based recommendations are:[4,6]
- Avoid high-dose vitamin C supplements. Vitamin C significantly boosts non-heme iron absorption. The amounts in food are fine, but supplemental doses above 250 mg per day should be avoided.[4,6]
- Limit alcohol. Alcohol amplifies iron-related liver damage and speeds up scarring. Patients with established liver disease should abstain. For others, low to moderate intake is the ceiling.[4,2]
- Avoid raw shellfish. Iron overload makes patients vulnerable to severe Vibrio vulnificus infection from raw oysters and other shellfish. This infection can be rapidly fatal. The risk holds until iron levels are normal.[6]
- Moderate red meat. Red meat contains heme iron, the most easily absorbed form. Moderation makes sense, but cutting it out completely is not supported by the evidence.[4]
- Tea or coffee with meals. These contain polyphenols that mildly reduce iron absorption. They are fine to drink with food.
Screening of Family Members
Every first-degree relative (parents, siblings, children) of a confirmed hemochromatosis patient should be offered cascade genetic testing and iron studies.[4,6] Siblings carry the highest genetic risk and should be tested first. Catching affected relatives before they sustain organ damage is the single most powerful prevention step in this condition. Children of an affected person only need genetic testing if the other parent is also a known carrier.
Ongoing Monitoring and Follow-Up
- Iron studies (TSAT and ferritin): every three to six months during induction, then every six to twelve months during maintenance.[4]
- Liver function tests at regular intervals.
- If cirrhosis was already present at diagnosis: liver ultrasound and serum AFP every six months for hepatocellular carcinoma surveillance. This continues even after iron levels normalize.[2,4]
- Assessment for diabetes, joint problems, and heart function in patients with significant iron loading.[7]
Prognosis
The outlook for hemochromatosis depends largely on two things: the stage at diagnosis and whether organ damage has already set in.[8]
Patients diagnosed before cirrhosis develops, and treated consistently with phlebotomy, have a life expectancy in line with the general population.[4,6] Fatigue and most blood-test abnormalities resolve once iron stores normalize. Heart changes and hormone disruption may partly or fully reverse.[7]
Patients who already have cirrhosis at diagnosis face a much worse outlook, no matter what treatment follows. The risk of liver cancer is around 200 times higher than the general population, and they also face the complications of portal hypertension.[2] Established joint disease and some forms of diabetes do not reverse with iron removal.[7]
This sharp split in outcomes — between pre-cirrhotic and cirrhotic patients at diagnosis — is exactly why early detection matters so much. Public awareness, family cascade testing, and opportunistic screening in primary care are the practical levers that move outcomes.
Living with Hemochromatosis
For patients diagnosed early, hemochromatosis is one of the most manageable chronic genetic disorders out there. With regular phlebotomy, most people lead normal, active lives. The treatment itself, blood removal, is straightforward and well tolerated. The hardest part is usually psychological: living with a lifelong condition that requires regular hospital visits. Specialist nursing support, patient advocacy groups (such as the Iron Disorders Institute in the USA), and counseling when needed can make a real difference to quality of life and treatment adherence.
Frequently Asked Questions (FAQs)
What are the different types of hereditary hemochromatosis?
Hemochromatosis is grouped into four main types, based on the gene involved.[3,4] Type 1 (HFE-related) is by far the most common. It is caused by mutations in the HFE gene and usually shows up in middle age. Type 2 (juvenile hemochromatosis) is caused by mutations in HJV or HAMP. It appears before age 30 and brings severe heart and hormone complications. Type 3 (TFR2-related) is rare and moderate in severity. Type 4 (ferroportin disease) is unusual: it is autosomal dominant, and it can cause a different pattern of iron loading, mainly in macrophages. See Table 1 for a comparison.
What is stage 1 of hemochromatosis?
Stage 1 is the phase of genetic susceptibility before any biochemical or clinical evidence of iron overload appears.[4] At this stage, blood tests, including transferrin saturation and ferritin, are normal. The person carries the causative mutations but has not yet built up detectable extra iron. Stage 1 can last years to decades. Catching it here, often through family cascade testing, gives the best possible window to act before any organ damage occurs.
What is the life expectancy of someone with hereditary hemochromatosis?
With early diagnosis, before cirrhosis or other significant organ damage, and with consistent phlebotomy, life expectancy matches the general population.[8] The landmark study by Niederau and colleagues (1996) showed that patients without cirrhosis at diagnosis had normal survival. Patients who present with established cirrhosis have a clearly reduced prognosis because of the elevated liver cancer risk and the chance of liver failure. This is the strongest argument for finding the disease early.[2,8]
Does hemochromatosis cause changes in stool color?
Stool color is not a reliable sign of hemochromatosis. Very high doses of iron supplements can darken the stool, but that is not a clinical feature of the disease itself. Dark or tarry stools (melena) point to bleeding higher up in the digestive tract and need urgent evaluation, regardless of hemochromatosis status. This is especially relevant for patients with cirrhosis and esophageal varices.
Can you take vitamin B12 if you have hemochromatosis?
Yes. Vitamin B12 does not increase iron absorption and is safe in hemochromatosis. The supplement to watch is vitamin C (ascorbic acid), which significantly boosts dietary iron absorption and should be avoided in supplemental doses.[4] Other B-complex vitamins and most minerals are also safe. Always tell the treating clinician about any supplements being taken so the full picture can be considered.
What is hereditary hemochromatosis and how is it different from ordinary iron deficiency?
Hereditary hemochromatosis (HH) is the opposite of iron deficiency. Rather than having too little iron, people with HH absorb too much iron from their food because of a fault in the gene (usually HFE) that regulates absorption. Over years, this excess iron builds up in organs — particularly the liver, heart, and pancreas — causing progressive damage. Iron deficiency anaemia causes fatigue and pallor due to too little iron; HH causes organ failure due to too much. Both conditions can involve fatigue, which is one reason HH can be missed.
If hemochromatosis runs in my family, does that mean I will definitely get it?
Not necessarily. HH follows an autosomal recessive inheritance pattern, meaning you need to inherit faulty copies of the HFE gene from both parents to be at risk. Even then, the condition has incomplete penetrance: studies suggest that only around 25–50% of people who inherit two copies of the most common mutation (C282Y) actually develop clinically significant iron overload. Factors including sex, alcohol intake, and diet influence whether symptoms develop. However, if a family member is diagnosed, all first-degree relatives (parents, siblings, children) should be offered genetic testing and iron studies, as early detection before organ damage occurs significantly improves outcomes.
How is the diagnosis confirmed, and when is a liver biopsy needed?
Diagnosis typically begins with two blood tests: transferrin saturation (TSAT) and serum ferritin. A TSAT above 45% on a fasting sample is the key initial red flag. This is followed by genetic testing for HFE mutations (C282Y and H63D). In most patients who are C282Y homozygotes with elevated iron markers, genetic testing alone is sufficient to confirm the diagnosis. Liver biopsy is now reserved for specific situations — for example, when the diagnosis remains uncertain, when the patient is not homozygous for C282Y, when ferritin is markedly elevated (typically above 1,000 µg/L), or when cirrhosis is suspected. MRI of the liver (T2* or R2* sequences) is a validated non-invasive alternative for estimating hepatic iron concentration.
What does treatment actually involve, and does it have to continue for life?
The main treatment is therapeutic phlebotomy — regular removal of blood, similar to blood donation. Each unit of blood removed contains approximately 200–250 mg of iron. Treatment begins with an intensive phase, often weekly sessions, until iron stores are depleted (ferritin falls below 50 µg/L and TSAT normalises). This can take months to over a year depending on iron burden. After the intensive phase, maintenance phlebotomy — typically three to four times per year — is required indefinitely to prevent iron re-accumulation. Most patients tolerate this well. In countries where it is permitted, therapeutic donations can be made to blood services. Chelation therapy with agents such as deferasirox is reserved for patients who cannot tolerate phlebotomy.
Will treatment reverse the damage already done to my liver or joints?
It depends on the extent of damage at diagnosis. If treatment begins before significant organ damage has occurred, most patients can expect a normal life expectancy and resolution of fatigue, liver enzyme abnormalities, and some cardiac and hormonal changes. However, certain complications — including established cirrhosis, arthropathy (joint damage), and some forms of diabetes — may not be reversible even after iron is depleted. This is why early diagnosis is so important. Patients with cirrhosis require ongoing surveillance for hepatocellular carcinoma (liver cancer) even after successful treatment, as their risk remains elevated.
Are there foods or supplements I need to avoid if I have hemochromatosis?
Dietary change is a supportive measure, not the primary treatment — phlebotomy is far more effective at removing iron than dietary restriction alone. That said, certain practices are recommended: avoid high-dose vitamin C supplements (vitamin C significantly enhances iron absorption); avoid raw shellfish, particularly oysters, as they carry a risk of severe Vibrio vulnificus infection in people with iron overload; and keep alcohol intake to a minimum, as alcohol worsens liver damage. Iron-fortified foods and red meat can be consumed in moderation; extreme dietary restriction is neither necessary nor recommended. Drinking tea or coffee with meals can mildly inhibit iron absorption and is generally acceptable.
Glossary of Related Medical Terms
- Autosomal recessive: A pattern of inheritance where a person must inherit two faulty copies of a gene — one from each parent — to develop the condition. People with only one faulty copy are called carriers and do not usually become unwell.
- Bronze diabetes: A historical term for the triad of skin bronzing, liver disease, and diabetes seen in advanced hemochromatosis. It is no longer used as a formal diagnosis but remains a useful clinical descriptor.
- Cardiomyopathy: Disease of the heart muscle. In hemochromatosis, iron deposits within the heart muscle weaken it, potentially causing heart failure or abnormal heart rhythms.
- Chelation therapy: Treatment using drugs (chelating agents) that chemically bind to iron in the bloodstream, allowing it to be excreted in urine. Used when regular blood removal is not possible.
- Cirrhosis: Severe scarring of the liver caused by long-term damage. Once cirrhosis develops, the scarring is irreversible, although stopping further damage with treatment can prevent progression.
- C282Y mutation: The most common genetic mutation causing hereditary hemochromatosis. At position 282 in the HFE protein, cysteine is replaced by tyrosine, disrupting the protein's ability to regulate iron absorption.
- Ferroportin: A protein that transports iron out of intestinal cells and macrophages into the bloodstream. It is regulated by hepcidin and is central to controlling how much iron enters circulation.
- Ferritin: A storage protein that holds iron within cells. Blood ferritin levels reflect total body iron stores; high ferritin can indicate iron overload but also occurs in inflammation and liver disease.
- H63D mutation: A secondary HFE gene mutation associated with a milder increase in iron absorption. It is more common than C282Y in the general population but causes significant disease mainly in combination with C282Y.
- Hepcidin: A small hormone produced by the liver that acts as the body's main iron regulator. It controls how much iron is absorbed from food and released from storage cells. In HH, hepcidin production is abnormally low, allowing unchecked iron absorption.
- Hepatocellular carcinoma (HCC): A type of primary liver cancer. Patients with hemochromatosis who develop cirrhosis have a significantly elevated risk of HCC, even after iron levels are normalised.
- HFE gene: A gene located on chromosome 6 that encodes a protein involved in regulating iron absorption. Mutations in this gene are the most common cause of hereditary hemochromatosis.
- Homozygous: Having two identical copies of a gene variant — one inherited from each parent. Most people with symptomatic HFE-related hemochromatosis are homozygous for the C282Y mutation.
- Hyperferritinaemia: Elevated levels of ferritin in the blood. While it can indicate iron overload, it is a non-specific finding also seen in obesity, metabolic syndrome, liver disease, alcohol use, and inflammatory conditions.
- Incomplete penetrance: When not everyone with the genetic change goes on to develop the disease. In HH, many people who are C282Y homozygotes never develop clinically significant iron overload.
- Iron overload (siderosis): Accumulation of excess iron in the body's organs and tissues. Mild overload may cause no symptoms; severe overload can damage the liver, heart, joints, and endocrine organs.
- Phlebotomy (venesection): The therapeutic removal of blood. In HH, regular blood removal is used to deplete excess iron from the body, as red blood cells contain iron-rich haemoglobin.
- Transferrin: A blood protein that transports iron around the body. Measuring how much transferrin is saturated with iron (transferrin saturation, or TSAT) is a key test for diagnosing hemochromatosis.
- Transferrin saturation (TSAT): A blood test showing what percentage of transferrin is carrying iron. A TSAT above 45% is the key early marker for suspected hemochromatosis.
- Vibrio vulnificus: A bacterium found in raw shellfish (particularly oysters) that can cause severe, potentially fatal infections in people with iron overload. Patients with HH are advised to avoid raw shellfish.
Disclaimer: This article is intended for informational purposes only and is specifically targeted towards medical students. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Adams, P. C., & Barton, J. C. (2007). Haemochromatosis. Lancet (London, England), 370(9602), 1855–1860. https://doi.org/10.1016/S0140-6736(07)61782-6
- Bacon, B. R., Adams, P. C., Kowdley, K. V., Powell, L. W., Tavill, A. S., & American Association for the Study of Liver Diseases (2011). Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology (Baltimore, Md.), 54(1), 328–343. https://doi.org/10.1002/hep.24330
- Brissot, P., Pietrangelo, A., Adams, P. C., de Graaff, B., McLaren, C. E., & Loréal, O. (2018). Haemochromatosis. Nature reviews. Disease primers, 4, 18016. https://doi.org/10.1038/nrdp.2018.16
- European Association for the Study of the Liver (2022). EASL Clinical Practice Guidelines on haemochromatosis. Journal of hepatology, 77(2), 479–502. https://doi.org/10.1016/j.jhep.2022.03.033
- Ganz T. (2013). Systemic iron homeostasis. Physiological reviews, 93(4), 1721–1741. https://doi.org/10.1152/physrev.00008.2013
- Kowdley, K. V., Brown, K. E., Ahn, J., & Sundaram, V. (2019). ACG Clinical Guideline: Hereditary Hemochromatosis. The American journal of gastroenterology, 114(8), 1202–1218. https://doi.org/10.14309/ajg.0000000000000315
- Murphree, C. R., Nguyen, N. N., Raghunathan, V., Olson, S. R., DeLoughery, T., & Shatzel, J. J. (2020). Diagnosis and management of hereditary haemochromatosis. Vox sanguinis, 115(4), 255–262. https://doi.org/10.1111/vox.12896
- Niederau, C., Fischer, R., Pürschel, A., Stremmel, W., Häussinger, D., & Strohmeyer, G. (1996). Long-term survival in patients with hereditary hemochromatosis. Gastroenterology, 110(4), 1107–1119. https://doi.org/10.1053/gast.1996.v110.pm8613000
- Pietrangelo A. (2010). Hereditary hemochromatosis: pathogenesis, diagnosis, and treatment. Gastroenterology, 139(2), 393–408.e4082. https://doi.org/10.1053/j.gastro.2010.06.013
- Porter JL, Rawla P. Hemochromatosis. [Updated 2024 Oct 6]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2026 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK430862/