Key Takeaways
Primary hemostasis is the body's rapid first response to a damaged blood vessel, forming a temporary platelet plug within minutes to stop bleeding before the coagulation cascade takes over.
- The Steps in Primary Hemostasis ▾: The four steps are vasoconstriction, platelet adhesion (via von Willebrand factor and collagen), platelet activation (shape change and granule release), and platelet aggregation (linked together by fibrinogen).
- Disorders of Primary Hemostasis ▾: Disorders fall into three groups: low platelet count (thrombocytopenia), poor platelet function (e.g., Glanzmann thrombasthenia, von Willebrand disease), and vessel wall defects (e.g., Ehlers-Danlos syndrome).
- Common Symptoms ▾: The classic bleeding pattern is mucocutaneous: petechiae, easy bruising, nosebleeds, gum bleeding, and heavy menstrual periods. Spontaneous bleeding usually appears once platelet counts drop below about 20 × 10⁹/L.
- General Investigations ▾: Modern workup includes a complete blood count, peripheral smear, PFA-100/200, light transmission aggregometry, and specific vWF assays. The older bleeding time test is largely retired [1,4].
- Treatment and Management ▾: Treatment depends on cause; platelet transfusion, desmopressin, antifibrinolytics (tranexamic acid), thrombopoietin receptor agonists for chronic ITP, rituximab, and caplacizumab for acquired TTP [5,7].
*Click ▾ for more information
Introduction
A paper cut stops bleeding in under five minutes. A patient on chemotherapy can bruise from a gentle squeeze. The difference is primary hemostasis. Understanding how the platelet plug forms and what disrupts it is the foundation for diagnosing bleeding disorders, interpreting coagulation panels, and using drugs like aspirin or clopidogrel safely.
This article walks through the steps in order, then maps them onto the disorders, investigations, and treatments you will see in clinical practice.
What is Primary Hemostasis?
Hemostasis is the set of processes that stop blood loss after vessel injury. It runs in two phases.
Primary hemostasis is the fast phase. Platelets stick to the injury site and clump together to form a soft plug, usually within 3 to 5 minutes [1].
Secondary hemostasis is the slower phase. Coagulation factors react in sequence to produce fibrin, which weaves through the platelet plug and locks it in place.
The two phases work together. In healthy vessels, the endothelial lining actively prevents clotting by releasing prostacyclin and nitric oxide, which keep platelets quiet [4]. Clotting only starts when this protective surface is breached.
The Four Steps of Primary Hemostasis
Step 1: Vasoconstriction
While traditionally taught as the first step of primary hemostasis, modern physiological models often classify vasoconstriction as a distinct initial "Vascular Phase." Similarly, Step 5 (Clot Retraction) is technically a post-coagulation event, as it relies heavily on the fibrin mesh generated during secondary hemostasis [4].
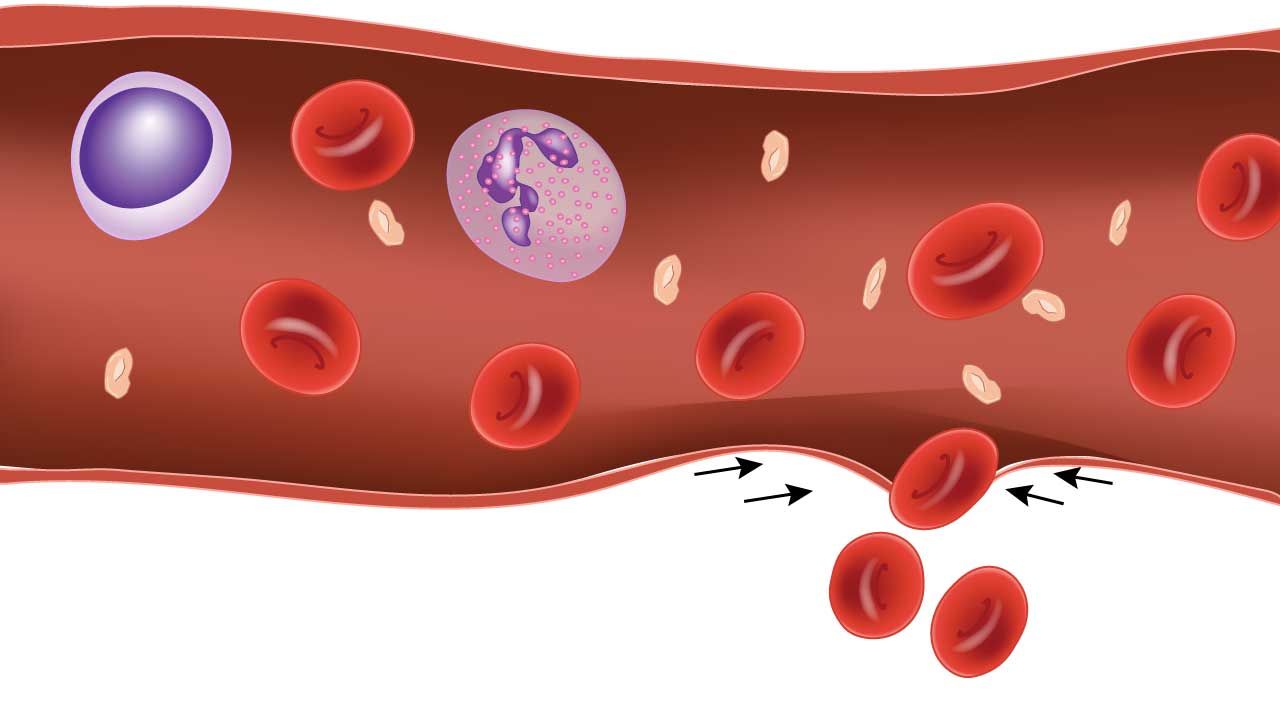
When a vessel is cut, the smooth muscle in its wall contracts within seconds. This narrows the vessel and reduces blood loss while platelets get to work.
Two signals drive the contraction. Endothelin, released by injured endothelial cells, binds smooth muscle receptors and triggers contraction through calcium-dependent pathways. Sympathetic reflexes add to this through norepinephrine release. Together, they buy time for the next steps in primary hemostasis [1,3].
Step 2: Platelet Adhesion
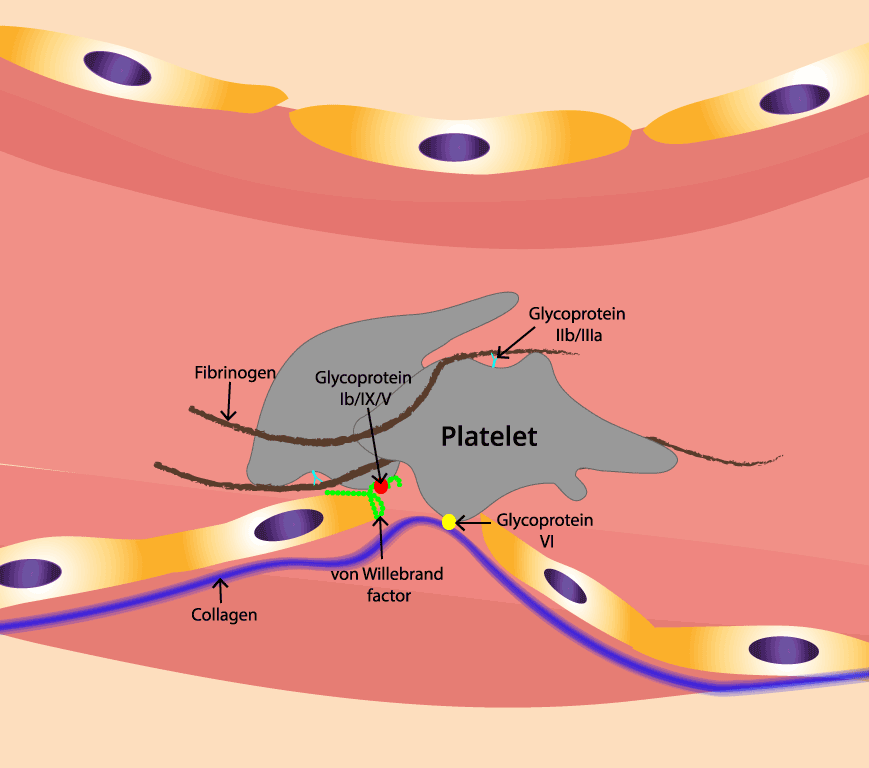
Injury exposes the collagen that sits beneath the endothelium. Platelets recognize this exposed collagen and stick to it. They do this in two ways.
Indirectly through von Willebrand factor (vWF). vWF is a large plasma protein produced by endothelial cells and megakaryocytes (the giant bone marrow cells that make platelets). At an injury site, blood flow stretches vWF and exposes its A1 domain. The A1 domain grabs the GPIbα receptor on passing platelets, tethering them to the wall.
Directly to collagen. The platelet receptor GPVI binds collagen itself and triggers strong activation signals. The integrin α2β1 reinforces this grip [3,4].
Red blood cells (RBCs) play a crucial rheological role in this process. Because they are larger and more deformable, RBCs flow in the center of the vessel, mechanically pushing platelets toward the vessel periphery (a process called margination) so they are perfectly positioned to interact with the damaged wall. Furthermore, under shear stress, RBCs release their own ADP to assist in early platelet activation [11].
The enzyme ADAMTS13 continuously trims ultra-large vWF strands in circulating blood into smaller, less sticky pieces. This prevents unwanted platelet clumping. When ADAMTS13 is deficient, ultra-large vWF accumulates and causes pathological clotting, which is the mechanism behind thrombotic thrombocytopenic purpura (TTP) [3].
Step 3: Platelet Activation
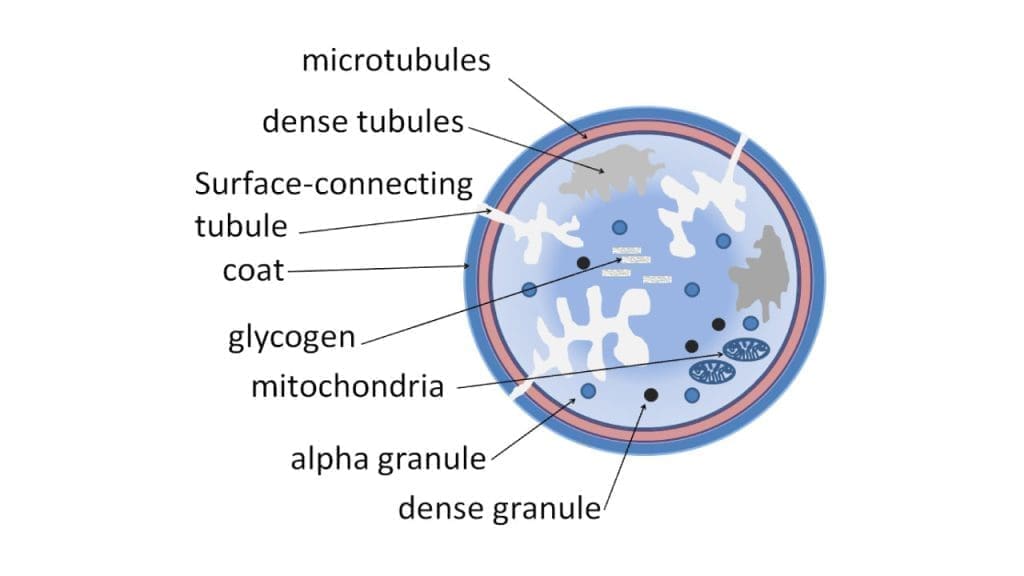
Once anchored, platelets transform. They change from smooth discs into spiky shapes that maximize their surface area, and they release the contents of their internal granules.
Dense granules release ADP, serotonin, and calcium. ADP recruits more platelets. Serotonin reinforces vasoconstriction. Calcium drives further activation.
Alpha granules release fibrinogen, vWF, P-selectin, and growth factors that support later wound healing.
Activated platelets also produce thromboxane A₂ (TxA₂) through the cyclooxygenase (COX) enzyme. TxA₂ is a strong amplifier of activation and vasoconstriction. This is why aspirin, which blocks COX, has antiplatelet effects [3,4].
A key consequence of activation is that the GPIIb/IIIa receptor on the platelet surface changes shape and becomes able to bind fibrinogen, which is the bridge for the next step.
Thrombin (while traditionally viewed as the end-product of secondary hemostasis) is generated locally in trace amounts almost immediately upon injury. Thrombin binds to Protease-Activated Receptors (PAR-1 and PAR-4) on the platelet surface, acting as the most potent platelet activator in the human body and illustrating the immense overlap between the primary and secondary hemostatic phases [12].
Step 4: Platelet Aggregation
Activated platelets link to each other through fibrinogen, which binds GPIIb/IIIa receptors on adjacent platelets. As more platelets arrive, they bind, activate, and release more ADP and TxA₂. This positive feedback rapidly builds a dense plug at the injury site.
vWF and P-selectin add stability. Once the plug is in place, the coagulation cascade weaves fibrin through it during secondary hemostasis [3].
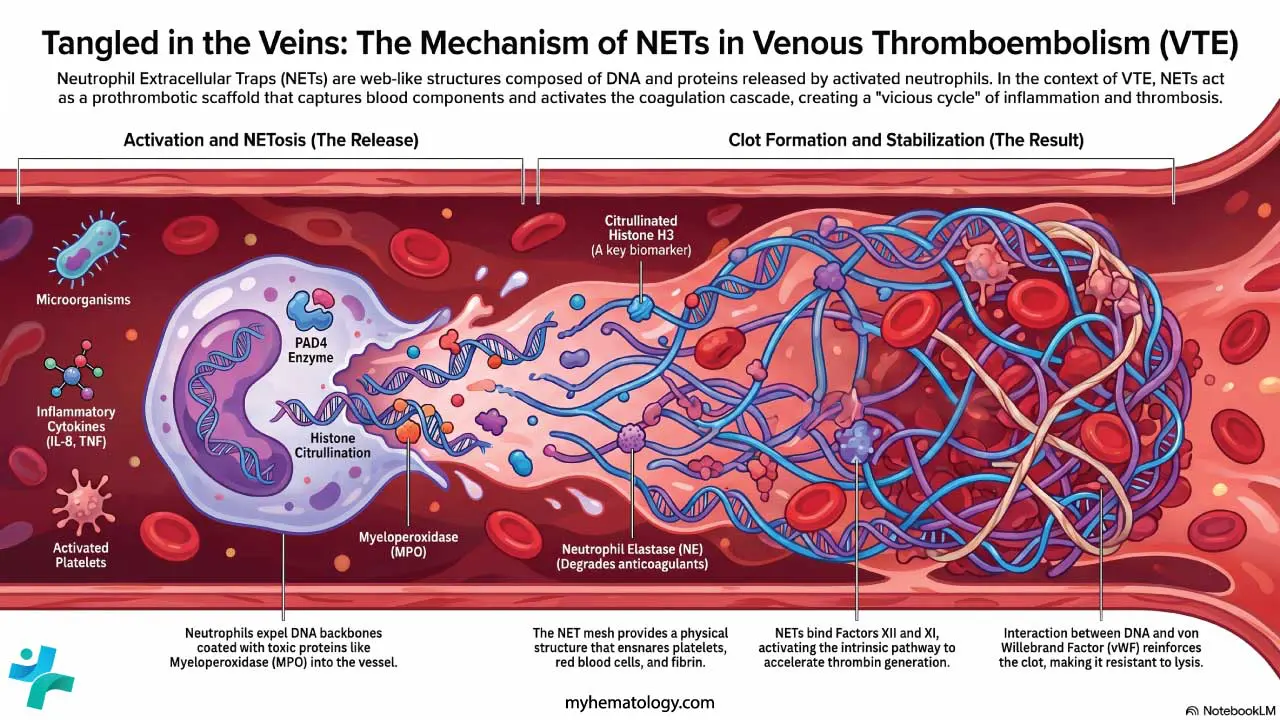
Modern hematology also heavily emphasizes the concept of thromboinflammation. Injured endothelium recruits immune cells, particularly neutrophils, which expel their DNA and histones to form Neutrophil Extracellular Traps (NETs). These sticky DNA webs capture platelets and red blood cells, providing a robust additional scaffold that reinforces the developing platelet plug [13].
Step 5: Clot Retraction
The plug is not the end. Platelets contract using their internal actin and myosin, pulling the wound edges closer and tightening the fibrin mesh. This shrinks the clot, restores partial blood flow in small vessels, and helps wound healing [4].
Disorders of Primary Hemostasis
Primary hemostasis can fail in three ways: too few platelets, dysfunctional platelets, or a damaged vessel wall.
Low Platelet Count (Thrombocytopenia)
Thrombocytopenia is a platelet count below 150 × 10⁹/L. Spontaneous bleeding risk rises sharply below 20 × 10⁹/L; surgical bleeding is a concern below about 50 × 10⁹/L [1,5].
Decreased production
- Bone marrow failure: aplastic anemia, myelodysplastic syndromes, leukemia.
- Marrow infiltration: metastatic cancer, fibrosis.
- Nutritional: vitamin B12 and folate deficiency.
- Drug-induced: chemotherapy agents.
Increased destruction
- Immune thrombocytopenia (ITP) — autoantibodies destroy platelets.
- Heparin-induced thrombocytopenia (HIT) — antibodies to heparin–platelet factor 4 complexes.
- Vaccine-induced immune thrombotic thrombocytopenia (VITT) — a rare reaction to adenoviral COVID-19 vaccines, recognized in 2021 [9].
- TTP — ADAMTS13 deficiency.
- Hemolytic uremic syndrome (HUS), disseminated intravascular coagulation (DIC), sepsis.
- Drug-induced — quinine, vancomycin, linezolid, and others.
Sequestration
- Splenomegaly from liver cirrhosis, portal hypertension, or hypersplenism.
Dysfunctional Platelets (Qualitative Disorders)
When platelets are present but cannot do their job, primary hemostasis still fails.
Adhesion defects
- Bernard-Soulier syndrome — GPIb/IX/V deficiency.
- Von Willebrand disease (VWD) — quantitative or qualitative vWF defect; the most common inherited bleeding disorder [8].
Activation defects
- Storage pool disorders — granule deficiencies.
- P2Y12 receptor defects — inherited or drug-induced (clopidogrel).
- Aspirin effect — irreversible COX inhibition.
Aggregation defects
- Glanzmann thrombasthenia — GPIIb/IIIa deficiency.
- Wiskott-Aldrich syndrome — defective cytoskeleton.
Vessel Wall Defects
Even with normal platelets, a faulty vessel wall can disrupt adhesion.
- Ehlers-Danlos syndrome — defective collagen.
- Hereditary hemorrhagic telangiectasia — fragile small vessels.
- Acquired — atherosclerosis, sepsis, burns, and radiation injury all damage the endothelium and impair vWF support.

How Primary Hemostasis Disorders Present
The bleeding pattern is the most useful clinical clue, and it is consistent across these disorders.
- Petechiae — pinpoint red or purple spots, usually on the lower legs.
- Purpura and ecchymoses — bruises, often spontaneous.
- Mucosal bleeding — nosebleeds, gum bleeding, heavy menstrual periods, GI bleeding, hematuria.
- Immediate bleeding after minor injury or dental work, often prolonged.
- Superficial rather than deep: joint bleeds (hemarthrosis) and large muscle hematomas point toward secondary hemostasis disorders like hemophilia.
Investigating Primary Hemostasis
A logical workup moves from screening tests to specific assays.
First-line tests
- Complete blood count (CBC) — platelet count and mean platelet volume (MPV). MPV is high in destructive thrombocytopenias and immune-related disorders, and low in marrow failure.
- Peripheral blood smear — confirms the count, identifies giant or small platelets (suggesting Bernard-Soulier or Wiskott-Aldrich), and rules out pseudo-thrombocytopenia from EDTA clumping.
- Coagulation screen (PT, aPTT) — usually normal in pure primary hemostasis disorders, which helps distinguish them from secondary hemostasis problems.
Functional tests
- PFA-100/200 (Platelet Function Analyzer) — a flow-based screening test that has largely replaced the bleeding time in modern labs [10].
- Light transmission aggregometry (LTA) — the reference test for platelet function, measuring response to ADP, collagen, ristocetin, arachidonic acid, and epinephrine.
- Whole-blood impedance aggregometry — used in some centers and in point-of-care settings.
Specific assays
- vWF antigen, vWF ristocetin cofactor activity, factor VIII level — for suspected VWD [8].
- ADAMTS13 activity — for suspected TTP.
- Flow cytometry — increasingly utilized alongside light transmission aggregometry in specialized hemostasis centers to map exact glycoprotein defects on the platelet surface (e.g., quantifying GPIIb/IIIa or GPIb expression) for highly tailored diagnostics [17].
- Genetic testing — for inherited platelet disorders.
The bleeding time test is mentioned for historical context but is rarely used today because of poor reproducibility [10].
Managing Primary Hemostasis Disorders
Treatment depends on cause, severity, and whether the patient is bleeding now or at risk of bleeding later.
Address the underlying cause
- Stop the offending drug.
- Treat infection or autoimmune trigger.
- Replace deficient nutrients (B12, folate, iron).
Control active bleeding
- Platelet transfusion for severe thrombocytopenia with bleeding or before procedures.
- Antifibrinolytics — tranexamic acid or aminocaproic acid stabilize clots by blocking plasmin. Useful in menorrhagia, epistaxis, and dental procedures.
- Desmopressin (DDAVP) raises endogenous vWF and factor VIII; helpful in mild type 1 VWD and some platelet function disorders [8].
Disease-specific therapies
- ITP — corticosteroids and IV immunoglobulin first-line; thrombopoietin receptor agonists (romiplostim, eltrombopag, avatrombopag) and rituximab are second-line options endorsed by the 2019 ASH guidelines [5]. Newer targeted therapies for refractory ITP have also entered routine clinical use. These include oral spleen tyrosine kinase (Syk) inhibitors like fostamatinib, which reduce the macrophage-mediated destruction of platelets, and neonatal Fc receptor (FcRn) inhibitors like efgartigimod, which rapidly degrade pathogenic IgG autoantibodies [14,15].
- Acquired TTP — plasma exchange plus immunosuppression; caplacizumab, an anti-vWF nanobody, was added to standard care after the HERCULES trial and is now part of guideline management [7]. Additionally, for patients with congenital TTP (cTTP), recombinant ADAMTS13 (apadamtase alfa) was recently FDA-approved as a targeted enzyme replacement therapy [16].
- VWD — desmopressin for mild cases; vWF/factor VIII concentrates or recombinant vWF (vonicog alfa) for severe disease [8].
- Inherited platelet function disorders — antifibrinolytics, platelet transfusion, and in selected severe cases, hematopoietic stem cell transplantation.
Frequently Asked Questions (FAQs)
What is primary hemostasis in simple terms?
Primary hemostasis is the body's first response to a cut or vessel injury. Within seconds, the vessel narrows and platelets stick to the injured wall, then clump together to form a soft plug that stops bleeding. It happens before the slower coagulation cascade builds the final fibrin clot.
How long does primary hemostasis take?
The platelet plug usually forms within 3 to 5 minutes of injury. Vasoconstriction starts almost immediately. The full process is rapid because it relies on platelets already circulating in the blood, not on proteins that need to be activated in sequence.
What is the difference between primary and secondary hemostasis?
Primary hemostasis forms a temporary platelet plug using platelets, von Willebrand factor, and exposed collagen. Secondary hemostasis follows and builds a stronger fibrin clot using coagulation factors. Disorders of primary hemostasis cause superficial bleeding (petechiae, gum bleeding, nosebleeds), while disorders of secondary hemostasis tend to cause deeper bleeding into joints and muscles.
Why does a low platelet count cause bleeding?
Platelets form the foundation of the initial plug. When the count drops below roughly 50 × 10⁹/L, surgical bleeding risk rises; below about 20 × 10⁹/L, spontaneous bleeding can occur. Without enough platelets, the body cannot build a plug fast enough at the site of injury, leading to easy bruising and mucosal bleeding.
Which lab tests assess primary hemostasis?
A complete blood count gives the platelet count and mean platelet volume. A peripheral blood smear shows platelet size and shape. The PFA-100/200 (platelet function analyzer) screens platelet function under flow. Light transmission aggregometry studies how platelets clump in response to specific agonists. Specific vWF antigen and activity assays diagnose von Willebrand disease. Bleeding time has largely been retired due to poor reproducibility.
What are common disorders of primary hemostasis?
Common conditions include immune thrombocytopenia (ITP), von Willebrand disease, drug-induced thrombocytopenia (heparin, quinine, vancomycin), thrombotic thrombocytopenic purpura (TTP), and inherited platelet function disorders such as Glanzmann thrombasthenia and Bernard-Soulier syndrome. Vaccine-induced immune thrombotic thrombocytopenia (VITT) is a rare but recognized cause linked to certain adenoviral vaccines.
Glossary of Related Medical Terms
- Primary hemostasis — the first stage of stopping blood loss, in which platelets form a temporary plug at the site of vessel injury.
- Platelet — a small, disc-shaped cell fragment in blood that helps form clots; produced by megakaryocytes in the bone marrow.
- von Willebrand factor (vWF) — a large plasma protein that bridges platelets to exposed collagen at injury sites.
- GPIbα — a glycoprotein receptor on platelets that binds vWF and starts platelet adhesion.
- GPIIb/IIIa (integrin αIIbβ3) — a platelet receptor that binds fibrinogen and links activated platelets together.
- Glycoprotein VI (GPVI) — a platelet receptor that binds directly to exposed collagen at injury sites.
- ADAMTS13 — an enzyme that cuts ultra-large vWF strands into smaller, less sticky pieces, preventing unwanted clotting.
- Thrombocytopenia — a low platelet count, usually below 150 × 10⁹/L (150,000/µL).
- Petechiae — pinpoint red or purple skin spots caused by bleeding from tiny vessels; classic sign of platelet disorders.
- Purpura — small bruises (under 1 cm) from bleeding into the skin.
- Ecchymosis — a larger bruise, generally over 1 cm.
- Mucocutaneous bleeding — bleeding from skin and mucous membranes (gums, nose, gut lining); the typical pattern in primary hemostasis disorders.
- Endothelium — the inner cell lining of blood vessels.
- Megakaryocyte — a giant bone marrow cell that sheds platelets into the bloodstream.
- Vasoconstriction — narrowing of a blood vessel by smooth muscle contraction.
- Thromboxane A₂ (TxA₂) — a lipid messenger released by activated platelets that promotes more platelet activation and vasoconstriction.
- PFA-100/200 — a modern bench-top test of platelet function under flow that has largely replaced bleeding time.
- Desmopressin (DDAVP) — a synthetic hormone that triggers vWF release from endothelial cells; used in mild von Willebrand disease.
- Thrombopoietin receptor agonist (TPO-RA) — a class of drugs (e.g., romiplostim, eltrombopag) that boost platelet production in chronic ITP.
- Caplacizumab — an anti-vWF nanobody used in acquired thrombotic thrombocytopenic purpura.
Disclaimer: This article is intended for educational and informational purposes only. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- LaPelusa A, Dave HD. Physiology, Hemostasis. [Updated 2023 May 1]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2026 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK545263/
- Periayah, M. H., Halim, A. S., & Mat Saad, A. Z. (2017). Mechanism Action of Platelets and Crucial Blood Coagulation Pathways in Hemostasis. International journal of hematology-oncology and stem cell research, 11(4), 319–327.
- Scridon A. (2022). Platelets and Their Role in Hemostasis and Thrombosis-From Physiology to Pathophysiology and Therapeutic Implications. International journal of molecular sciences, 23(21), 12772. https://doi.org/10.3390/ijms232112772
- Versteeg, H. H., Heemskerk, J. W., Levi, M., & Reitsma, P. H. (2013). New fundamentals in hemostasis. Physiological reviews, 93(1), 327–358. https://doi.org/10.1152/physrev.00016.2011
- Neunert, C., Terrell, D. R., Arnold, D. M., Buchanan, G., Cines, D. B., Cooper, N., Cuker, A., Despotovic, J. M., George, J. N., Grace, R. F., Kühne, T., Kuter, D. J., Lim, W., McCrae, K. R., Pruitt, B., Shimanek, H., & Vesely, S. K. (2019). American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood advances, 3(23), 3829–3866. https://doi.org/10.1182/bloodadvances.2019000966
- Liebman H. A. (2014). Thrombocytopenia in cancer patients. Thrombosis research, 133 Suppl 2, S63–S69. https://doi.org/10.1016/S0049-3848(14)50011-4
- Scully, M., Cataland, S. R., Peyvandi, F., Coppo, P., Knöbl, P., Kremer Hovinga, J. A., Metjian, A., de la Rubia, J., Pavenski, K., Callewaert, F., Biswas, D., De Winter, H., Zeldin, R. K., & HERCULES Investigators (2019). Caplacizumab Treatment for Acquired Thrombotic Thrombocytopenic Purpura. The New England journal of medicine, 380(4), 335–346. https://doi.org/10.1056/NEJMoa1806311
- Connell, N. T., Flood, V. H., Brignardello-Petersen, R., Abdul-Kadir, R., Arapshian, A., Couper, S., Grow, J. M., Kouides, P., Laffan, M., Lavin, M., Leebeek, F. W. G., O'Brien, S. H., Ozelo, M. C., Tosetto, A., Weyand, A. C., James, P. D., Kalot, M. A., Husainat, N., & Mustafa, R. A. (2021). ASH ISTH NHF WFH 2021 guidelines on the management of von Willebrand disease. Blood advances, 5(1), 301–325. https://doi.org/10.1182/bloodadvances.2020003264
- Greinacher, A., Thiele, T., Warkentin, T. E., Weisser, K., Kyrle, P. A., & Eichinger, S. (2021). Thrombotic Thrombocytopenia after ChAdOx1 nCov-19 Vaccination. The New England journal of medicine, 384(22), 2092–2101. https://doi.org/10.1056/NEJMoa2104840
- Godby, R. C., et al. (2022). Congenital and acquired disorders of primary hemostasis. Annals of Blood, 7, 10.
- Walton, B. L., Lehmann, M., Skorczewski, T., Holle, L. A., Beckman, J. D., Cribb, J. A., Mooberry, M. J., Wufsus, A. R., Cooley, B. C., Homeister, J. W., Pawlinski, R., Falvo, M. R., Key, N. S., Fogelson, A. L., Neeves, K. B., & Wolberg, A. S. (2017). Elevated hematocrit enhances platelet accumulation following vascular injury. Blood, 129(18), 2537–2546. https://doi.org/10.1182/blood-2016-10-746479
- Koupenova, M., Kehrel, B. E., Corkrey, H. A., & Freedman, J. E. (2017). Thrombosis and platelets: an update. European heart journal, 38(11), 785–791. https://doi.org/10.1093/eurheartj/ehw550
- Engelmann, B., & Massberg, S. (2013). Thrombosis as an intravascular effector of innate immunity. Nature reviews. Immunology, 13(1), 34–45. https://doi.org/10.1038/nri3345
- Bussel, J., Arnold, D. M., Grossbard, E., Mayer, J., Treliński, J., Homenda, W., Hellmann, A., Windyga, J., Sivcheva, L., Khalafallah, A. A., Zaja, F., Cooper, N., Markovtsov, V., Zayed, H., & Duliege, A. M. (2018). Fostamatinib for the treatment of adult persistent and chronic immune thrombocytopenia: Results of two phase 3, randomized, placebo-controlled trials. American journal of hematology, 93(7), 921–930. https://doi.org/10.1002/ajh.25125
- Broome, C., Miyakawa, Y., Carpenedo, M., Al-Samkari, H., Ayguasanosa, J., Phillips, J., & Rodeghiero, F. (2026). Efgartigimod as a treatment for adults with primary immune thrombocytopenia: a plain language summary of the ADVANCE IV study. Therapeutic advances in hematology, 17, 20406207261445668. https://doi.org/10.1177/20406207261445668
- Scully, M., Antun, A., Cataland, S. R., Coppo, P., Dossier, C., Biebuyck, N., Hassenpflug, W. A., Kentouche, K., Knöbl, P., Kremer Hovinga, J. A., López-Fernández, M. F., Matsumoto, M., Ortel, T. L., Windyga, J., Bhattacharya, I., Cronin, M., Li, H., Mellgård, B., Patel, M., Patwari, P., … cTTP Phase 3 Study Investigators (2024). Recombinant ADAMTS13 in Congenital Thrombotic Thrombocytopenic Purpura. The New England journal of medicine, 390(17), 1584–1596. https://doi.org/10.1056/NEJMoa2314793
- Frelinger A. L., 3rd (2024). Flow Cytometry and Platelets. Clinics in laboratory medicine, 44(3), 511–526. https://doi.org/10.1016/j.cll.2024.04.011