TL;DR
Chronic Myeloid Leukemia (CML) is a clonal myeloproliferative neoplasm characterized by the excessive and unregulated proliferation of mature and maturing granulocytic cells, primarily affecting the myeloid lineage.
- Hypermetabolism e.g. weight loss, anorexia or night sweats
- Splenomegaly
- Anemia, bruising, epistaxis or menorrhagia
- Hyperuricemia
- PBF: Normochromic normocytic anemia and leukocytosis
- BMA: Hypercellular with granulopoietic predominance
- NAP score: Low
- Cytogenetic analysis or qPCR: Presence of Philadelphia chromosome
- ↑ serum acid
- Tyrosine kinase inhibitors (TKIs)
- Stem cell transplantation
*Click ▾ for more information
What is Chronic Myeloid Leukemia (CML)?
Chronic myeloid leukemia (CML) is classified as a clonal hematopoietic stem cell disorder and is part of the broader group of myeloproliferative neoplasms (MPNs). The disease originates in a single, pluripotent hematopoietic stem cell that has undergone a critical genetic mutation. This cell gives rise to a massive, uncontrolled proliferation of progeny, primarily in the myeloid lineage.
The defining characteristic of chronic myeloid leukemia (CML) is the excessive production and accumulation of myeloid cells, granulocytes (neutrophils, eosinophils, and basophils), at all stages of maturation in the bone marrow and peripheral blood.
In its initial and most common presentation (Chronic Phase), chronic myeloid leukemia (CML) progresses slowly over months to years, which differentiates it from the aggressive course of Acute Leukemias.
Epidemiology and Incidence
Chronic myeloid leukemia (CML) is a relatively uncommon disease, accounting for approximately 15% of all leukemias in adults.
Age of Onset: While it can occur at any age, chronic myeloid leukemia (CML) is primarily a disease of older adults. The median age at diagnosis typically falls between 60 and 65 years. There is a slight male predominance.
Prevalence: The annual incidence is estimated to be around 1 to 2 cases per 100,000 adults.
What causes chronic myeloid leukemia (CML)?
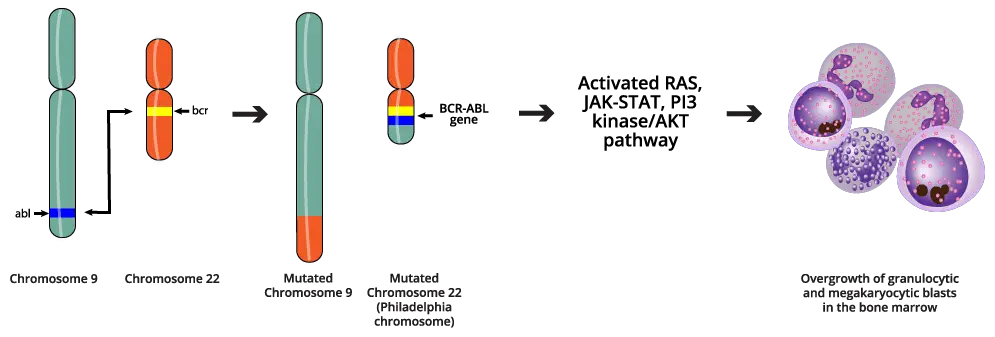
The fundamental cause of chronic myeloid leukemia (CML) is a single, specific, recurrent genetic event: the formation of the BCR-ABL1 fusion oncogene. This aberrant gene product drives the malignant transformation of the hematopoietic stem cell.
The Philadelphia (Ph) Chromosome
The genetic abnormality that defines chronic myeloid leukemia (CML) is the Philadelphia (Ph) chromosome, named for the city where it was first discovered. The Ph chromosome is the result of a reciprocal translocation between the long arms of chromosome 9 and chromosome 22. This event is designated as t(9;22)(q34;q11).
The long arm of chromosome 9 (9q34) contains the ABL1 (Abelson murine leukemia viral oncogene homolog 1) gene, a proto-oncogene that is normally involved in cellular signaling and the long arm of chromosome 22 (22q11) contains the BCR (Breakpoint Cluster Region) gene, a gene with regulatory functions.
The Resulting Chromosomes
The smaller, derivative chromosome 22 (der(22)) that contains the fused BCR-ABL1 gene is the Ph chromosome. This is the pathogenic chromosome. The ABL1 gene encodes a non-receptor tyrosine kinase. In its normal state, its activity is tightly regulated and only switched on in response to specific growth signals. However, the BCR component causes the new BCR-ABL1 protein to spontaneously form homodimers or oligomers. This dimerization forces the ABL1 kinase domain into a permanent, “on” or activated conformation.
The result is a BCR-ABL1 protein that possesses constitutively active tyrosine kinase activity, meaning it constantly phosphorylates downstream targets, regardless of external signals.
A larger, derivative chromosome 9 (der(9)) is also formed, but this product is generally thought to be non-pathogenic in chronic myeloid leukemia (CML).
Mechanism of Leukemogenesis
The perpetually active BCR-ABL1 kinase acts as a master regulator, hijacking multiple intracellular signaling pathways to create a favorable environment for malignancy.
The genomic instability caused by BCR-ABL1 sets the stage for clonal evolution. Over time, the leukemic clone acquires additional mutations (e.g., trisomy 8, isochromosome 17q), leading to genetic instability, loss of differentiation capacity, and ultimately, progression from the indolent Chronic Phase (CP) to the highly aggressive Accelerated Phase (AP) and Blast Crisis (BC).
BCR-ABL1 activates key signaling cascades, including the RAS/MAPK, PI3K/AKT, and JAK/STAT pathways. These signals bypass normal growth controls, promoting rapid and sustained proliferation of the myeloid lineage cells. The activation of the PI3K/AKT pathway leads to the suppression of programmed cell death (apoptosis). This extends the lifespan of the chronic myeloid leukemia (CML) cells, allowing them to accumulate in the bone marrow and peripheral blood.
BCR-ABL1 disrupts normal cell adhesion mechanisms, allowing immature cells to spill out of the bone marrow into the circulation. It also contributes to genomic instability, impairing DNA repair and increasing the rate of secondary mutations.
Chronic Myeloid Leukemia (CML) Clinical Presentation & Phases
Chronic myeloid leukemia (CML) is characterized by its natural history of a relatively indolent, chronic phase that invariably progresses to a fatal acute phase (Blast Crisis) if left untreated. Modern therapy with Tyrosine Kinase Inhibitors (TKIs) aims to keep the patient permanently in the Chronic Phase.
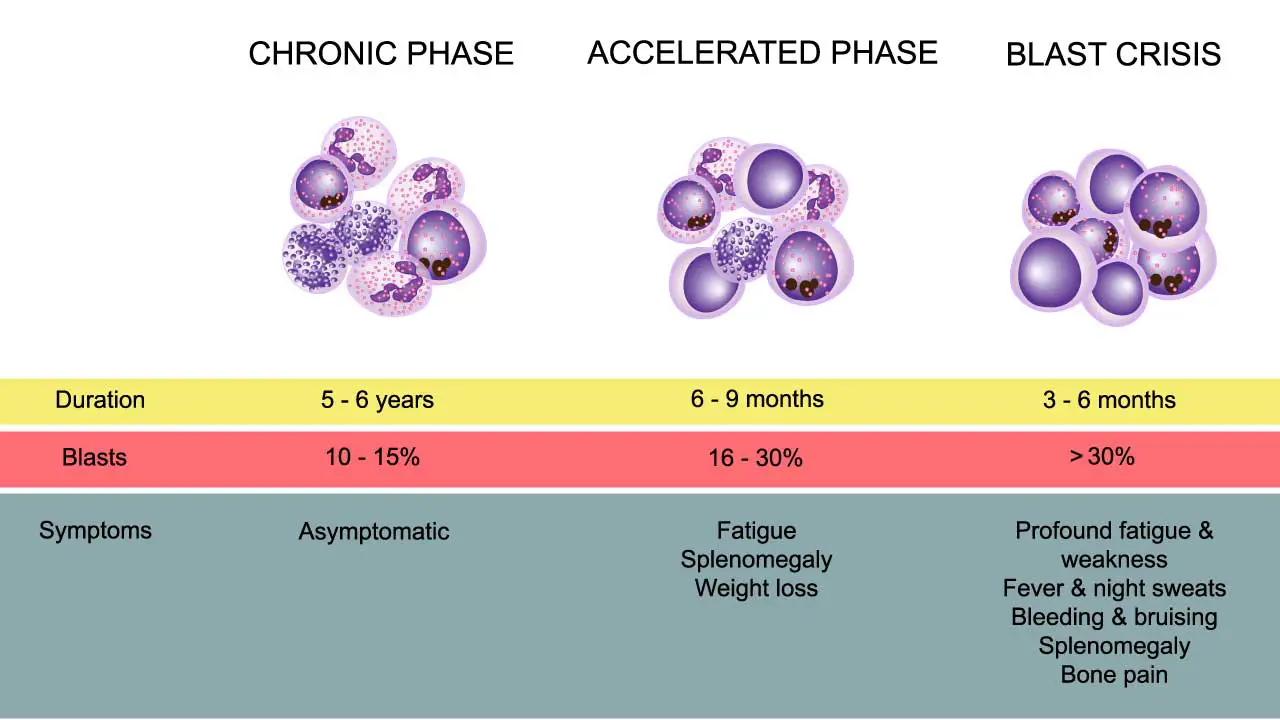
Chronic Phase (CP)
The Chronic Phase is the most common presentation, with 85−90% of patients diagnosed at this stage. It is characterized by the uncontrolled, yet well-differentiated, proliferation of myeloid cells.
Symptoms
Symptoms are often nonspecific, mild, or absent entirely (incidental finding on routine blood work).
- Constitutional Symptoms: Fatigue, low-grade fever, night sweats, and unintentional weight loss (common symptoms related to high cellular turnover and metabolic demand).
- Abdominal Fullness/Pain: Due to massive splenomegaly.
- Bleeding/Bruising: Due to functional platelet defects, even when platelet counts are high.
- Gout/Arthralgia: Related to increased uric acid production (hyperuricemia) from high cell turnover.
Physical Examination
- Splenomegaly: The most consistent and important physical finding. The spleen is often significantly enlarged, firm, and palpable below the costal margin.
- Hepatomegaly: Mild to moderate liver enlargement may be present.
- Signs of Hyperviscosity: Rarely, very high white blood cell (WBC) counts (≥200×109/L) can lead to hyperviscosity symptoms (priapism, visual disturbances, or central nervous system signs), requiring urgent management.
Accelerated Phase (AP)
The Accelerated Phase represents a phase of clonal evolution and disease instability, serving as a biological bridge between the Chronic Phase and Blast Crisis. The disease becomes more difficult to control with standard therapy. The specific criteria for defining the AP often vary slightly across organizations (e.g., WHO, ELN), but generally include the following features:
- Increasing Blasts: Peripheral blood or bone marrow blasts between 10% and 19%.
- Persistent Cytopenias: Unexplained, persistent thrombocytopenia (≤100×109/L) or persistent thrombocytopoiesis, unresponsive to therapy.
- Persistent Leukocytosis: Increasing WBC count despite treatment.
- Increasing Basophils: Peripheral blood basophils ≥20%.
- New Clonal Evolution: The development of new cytogenetic abnormalities (e.g., trisomy 8, isochromosome 17q, second Ph chromosome) in Ph-positive cells.
- Extramedullary Disease: New or enlarging soft tissue masses (granulocytic sarcomas).
Blast Crisis (BC)
Blast Crisis is the final and most aggressive stage of chronic myeloid leukemia (CML), representing transformation to acute leukemia. It carries a grave prognosis and is much harder to treat than the initial Chronic Phase.
- Defining Criteria: The most critical criterion for blast crisis is:
- Blasts ≥20% in the peripheral blood or bone marrow.
- Presence of extramedullary blast proliferation (granulocytic sarcoma).
- Lineage of Transformation: The transformation can follow two main paths:
- Myeloid Blast Crisis (MBC): The malignant clone differentiates into acute myeloid leukemia (AML). This is the most common form (60−70% of cases).
- Lymphoid Blast Crisis (LBC): The malignant clone differentiates into B-cell or T-cell acute lymphoblastic leukemia (ALL). This occurs in about 20−30% of cases and may respond better to ALL-specific chemotherapy regimens in conjunction with TKIs.
- Clinical Implications: Symptoms often resemble aggressive acute leukemia (severe cytopenias, infection, bleeding) and require immediate, intensive therapy.
How is chronic myeloid leukemia (CML) investigated?
The diagnosis of chronic myeloid leukemia (CML) requires a multi-modal approach, combining standard hematology tests with advanced molecular and cytogenetic studies to confirm the presence of the BCR-ABL1 oncogene.
Peripheral Blood Smear (PBF) and Complete Blood Count (CBC)
The initial findings from a routine blood test are often highly suggestive of CML.
- Leukocytosis: There is typically a marked and persistent elevation of the White Blood Cell (WBC) count in the complete blood count, often ranging from 25×109/L to over 500×109/L.
- Myeloid Left Shift: The blood smear shows a characteristic full spectrum of myeloid maturation (the myeloid bulge). This means that cells at almost every stage of granulocyte development are present, including myeloblasts, promyelocytes, myelocytes, metamyelocytes, bands, and segmented neutrophils. The number of myelocytes and neutrophils usually dominates.
- Key Morphological Clues:
- Basophilia: An absolute increase in basophils is a highly characteristic and important finding in chronic myeloid leukemia (CML).
- Eosinophilia: Eosinophil counts are also frequently elevated.
- Thrombocytosis: Platelet counts are often normal or elevated (thrombocytosis) in the Chronic Phase, though they can become low (thrombocytopenia) in the Accelerated Phase.
- Leukocyte Alkaline Phosphatase (LAP) Score: The LAP score, which measures an enzyme present in mature neutrophils, is characteristically low or absent in CML. This contrasts sharply with other causes of leukocytosis (like infection or leukemoid reaction), where the LAP score is high.
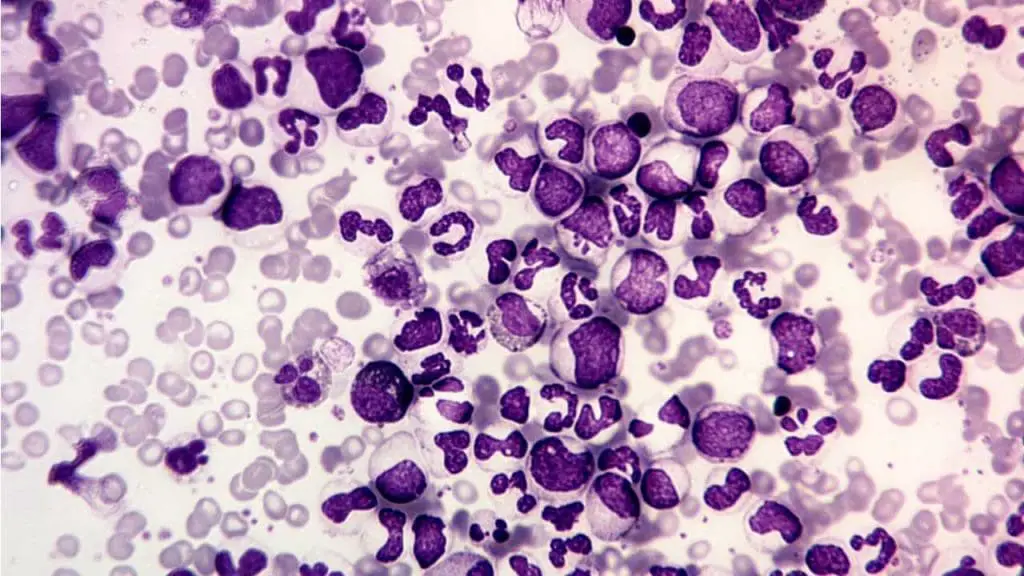
Bone marrow aspiration and biopsy
While the diagnosis is molecular, bone marrow examination provides information on cellularity, maturation, and ruling out progression.
- Cellularity: The marrow is usually markedly hypercellular (>80% cellularity).
- Granulocytic Hyperplasia: The primary feature is extreme hyperplasia of the granulocytic lineage, with an increased myeloid-to-erythroid (M:E) ratio, typically 10:1 to 30:1 (normal is 2:1 to 4:1).
- Megakaryocytes: Small, dysplastic megakaryocytes are often increased in number.
- Fibrosis: Reticulin fibrosis may be present, which can indicate a poorer prognosis or progression to the Accelerated Phase.
- Blast Count: The percentage of blasts is determined here to classify the disease phase:
- Chronic Phase (CP): Blasts <10%
- Accelerated Phase (AP): Blasts 10% to 19%
Blast Crisis (BC): Blasts ≥20%
- Chronic Phase (CP): Blasts <10%
Molecular and Cytogenetic Confirmation (Crucial for Definitive Diagnosis)
The definitive diagnosis relies on confirming the t(9;22) translocation and the presence of the BCR-ABL1 fusion gene.
Conventional Karyotyping (Cytogenetics)
Cytogenetics is essential for the initial diagnosis, as it can detect the primary Ph chromosome, t(9;22)(q34;q11), and any additional chromosomal abnormalities (clonal evolution) that signal progression (e.g., trisomy 8).
Fluorescence in situ Hybridization (FISH)
FISH technique uses fluorescently labeled DNA probes specific to the BCR and ABL1 regions by to detect the fusion signal (one red and one green signal merge to form a yellow fusion signal). This technique is useful for rapid confirmation in peripheral blood or when bone marrow cells fail to grow in culture for karyotyping. It is sensitive enough to detect the BCR-ABL1 fusion in up to 95% of CML cases.
Quantitative Polymerase Chain Reaction (qPCR)
This is the gold standard for monitoring treatment response. qPCR measures the reduction in the leukemic burden relative to a control gene and is reported on the International Scale (IS). This allows comparison of results globally and guides treatment decisions, particularly regarding goals like Major Molecular Response (MMR) and Treatment-Free Remission (TFR).
Prognostic Scoring and Risk Stratification
Prognostic scoring systems in chronic myeloid leukemia (CML) are designed to predict a patient’s long-term outcome before they start Tyrosine Kinase Inhibitor (TKI) therapy. These scores, based on easily obtainable clinical and hematological parameters at the time of diagnosis, help clinicians stratify patients into risk groups (low, intermediate, or high) to guide the selection of the most appropriate initial TKI.
The Traditional Sokal Risk Score
The Sokal Score was the first widely used prognostic tool for chronic myeloid leukemia (CML). It was developed in the pre-TKI era (when Interferon-alpha and chemotherapy were the standard treatments) to predict the median survival time for patients. While still sometimes mentioned, its use has largely been superseded by modern scores that are more accurate in the context of TKI therapy.
| Parameter | Contribution |
|---|---|
| Age | Older age is associated with worse prognosis. |
| Spleen Size | Significant splenomegaly indicates higher disease burden. |
| Platelet Count | Very high or very low platelet counts predict poorer outcomes. |
| Peripheral Blood Blasts | Higher blast percentage is the strongest negative predictor. |
Patients are categorized into low, intermediate, or high-risk groups based on a derived score calculated using a complex formula incorporating these four variables.
The EUTOS Long-Term Survival (ELTS) Score
The ELTS Score is the preferred modern risk stratification tool for chronic myeloid leukemia (CML) patients starting first-line TKI therapy (specifically, Imatinib). It was developed and validated using data from patients treated with Imatinib and is considered a better predictor of long-term outcomes in the modern treatment landscape, particularly Failure-Free Survival (FFS).
Components of the ELTS Score
The ELTS score uses the same four clinical parameters as the Sokal score but assigns them different weightings, reflecting the understanding that age is a more dominant factor in the TKI era.
| Parameter | Weighting/Threshold |
|---|---|
| Age | Scored differently for patients <50, 50−60, and >60 years. |
| Spleen Size | Scored by the number of cm below the costal margin. |
| Platelet Count | Scored based on absolute values (e.g., <67.5 or >434×109/L). |
| Peripheral Blood Blasts | Score assigned based on the percentage of blasts. |
ELTS Risk Groups and Interpretation
The final ELTS score places the patient into one of three risk categories:
- Low Risk (LR): Predicts excellent prognosis and high rate of long-term molecular response.
- Intermediate Risk (IR): Suggests a slightly increased risk of failing to achieve optimal response.
- High Risk (HR): Predicts the highest rate of treatment failure and progression.
Clinical Application of Risk Stratification
The primary utility of these scores is in guiding the initial selection of the TKI.
- Low/Intermediate Risk Patients: These patients generally have excellent outcomes with first-generation TKIs (Imatinib), which is often preferred due to its lower cost, established safety profile, and minimal non-hematological toxicities.
- High-Risk Patients: For patients classified as High Risk by ELTS, starting treatment with a second-generation TKI (e.g., Dasatinib or Nilotinib) is generally recommended. These agents are more potent and achieve deep molecular response (DMR) faster, which may mitigate the initial poor prognostic factors and improve FFS.
- Monitoring: Regardless of the initial score, strict adherence to the 3-month benchmark (achieving BCR-ABL1≤10% IS) remains the most crucial predictor of long-term success. If this benchmark is missed, treatment must be escalated, even in a previously low-risk patient.
Treatment and Management for CML
The management of chronic myeloid leukemia (CML) has been revolutionized by targeted therapy, transforming a uniformly fatal disease into a chronic, manageable condition for most patients. The goal is deep and sustained molecular remission. A deep dive into the current treatment and management of CML can be found in another article titled ‘Chronic Myeloid Leukemia Treatment Strategies‘.
Tyrosine Kinase Inhibitors (TKIs)
TKIs are the primary treatment for all phases of chronic myeloid leukemia (CML), especially the Chronic Phase. Their mechanism is direct: they competitively bind to the adenosine triphosphate (ATP)-binding pocket of the BCR-ABL1 fusion protein, preventing the constitutive tyrosine kinase activity and thus blocking the malignant signaling cascade.
Treatment Goals and Milestones
Treatment response is monitored using qPCR on the International Scale (IS) and is defined by several sequential milestones.
| Response Type | Definition (Goal) | Description |
|---|---|---|
| Hematologic Response (CHR) | Normalization of CBC | Normal WBC and platelet counts; no splenomegaly; WBC differential without immature granulocytes. |
| Cytogenetic Response (CCyR) | Elimination of Ph chromosome | No Ph chromosome detected via conventional karyotyping (less than 1% Ph-positive cells). |
| Major Molecular Response (MMR) | BCR-ABL1 ≤0.1% IS | BCR-ABL1 transcripts are reduced to ≤0.1% relative to the baseline standardized value. The key therapeutic goal. |
| Deep Molecular Response (DMR) | BCR-ABL1 ≤0.01% IS | Often referred to as MR4 (MMR with a 10-fold deeper reduction). Required before considering Treatment-Free Remission (TFR). |
Generations of TKIs![]()
The choice of TKI often depends on prognostic risk scores (e.g., Sokal, EUTOS), patient comorbidities, and the specific phase of the disease.
| Generation | Drug (Examples) | Key Feature / Rationale for Use |
|---|---|---|
| 1st Gen | Imatinib (Gleevec) | First-in-class, highly effective, generally well-tolerated. Still a standard first-line choice, especially for low-risk patients or those with cardiovascular risk factors. |
| 2nd Gen | Dasatinib, Nilotinib | More potent than Imatinib, achieving faster and deeper responses. Used first-line, especially in high-risk patients, or second-line for Imatinib resistance/intolerance. |
| 3rd Gen | Ponatinib, Bosutinib | Designed to overcome most common TKI resistance mutations. Ponatinib is effective against the notorious T315I mutation. Reserved for highly refractory or resistant disease. |
| 4th Gen | Asciminib (ABL-specific) | Binds to a different site (the myristoyl pocket) of ABL, providing unique efficacy against certain resistant clones (excluding T315I) and often reserved for third-line therapy. |
Monitoring and Management of Therapy
Treatment with TKIs is lifelong unless the patient meets criteria for TFR. Rigorous monitoring is mandatory.
- Monitoring Schedule: qPCR should be performed every 3 months until MMR is achieved, and then every 3−6 months thereafter to ensure stability.
- Response Benchmarks: The European LeukemiaNet (ELN) guidelines provide clear benchmarks for optimal response. Failure to meet these benchmarks often requires switching to a different, more potent TKI. Failure to Achieve BCR-ABL1≤10% IS at 3 months is a critical predictor of poor long-term outcome and usually triggers a change in therapy.
- Addressing Resistance: Most resistance occurs due to point mutations in the ABL1 kinase domain, preventing the TKI from binding.
- The T315I Mutation: This is the most challenging mutation, as it creates a steric hindrance that prevents binding of all TKIs except Ponatinib (and potentially the new 4th generation agent, Asciminib, which binds an allosteric pocket). Mutational analysis must be performed upon confirmed treatment failure.
Treatment-Free Remission (TFR)
TFR is the major therapeutic goal of the current era, allowing a patient to safely stop TKI therapy without disease recurrence.
- Eligibility Criteria: Patients must have:
- Durable DMR (MR4 or MR4.5) for a minimum of 2−5 years.
- No history of chronic myeloid leukemia (CML) progression (AP or BC).
- Treatment should have been continuous and consistent.
- Post-Cessation Monitoring: Patients who stop TKI therapy must be monitored very closely (monthly qPCR for the first 6−12 months).
- Relapse and Re-treatment: If molecular relapse occurs (e.g., loss of MMR), the TKI is immediately restarted. Re-treatment with the original TKI is highly effective in regaining MMR.
Role of Allogeneic Hematopoietic Stem Cell Transplantation (ASCT)
Before TKIs, ASCT was the only curative option. Today, its role is highly restricted due to high morbidity and mortality.
Current Indications: ASCT is typically reserved for:
- Primary or secondary resistance to multiple TKIs (especially in patients who fail Ponatinib).
- Patients presenting in or progressing to Accelerated Phase (AP) or Blast Crisis (BC) where TKI effectiveness is limited.
- Patients with difficult-to-treat mutations, where TKI options have been exhausted.

Frequently Asked Questions (FAQs)
What is CML life expectancy?
CML life expectancy has significantly improved due to advancements in treatment, particularly tyrosine kinase inhibitors (TKIs). With effective treatment, many patients with CML can live long, near-normal lives. However, factors such as the stage of the disease at diagnosis, response to treatment, and the development of resistance can influence individual outcomes. Regular monitoring and adherence to treatment plans are crucial for managing CML and maximizing life expectancy.
Can CML be cured?
While there is no definitive cure for chronic myeloid leukemia (CML), modern treatments, particularly tyrosine kinase inhibitors (TKIs), have significantly improved outcomes. Many patients with chronic myeloid leukemia (CML) can live long, near-normal lives. With ongoing advancements in research and treatment, the goal of curing chronic myeloid leukemia (CML) remains a focus for medical professionals.
What are the three stages of CML?
Chronic myeloid leukemia (CML) is typically divided into three phases.
- Chronic phase: This is the initial stage, characterized by a gradual increase in abnormal white blood cells. Symptoms may be mild or absent during this phase.
- Accelerated phase: As the disease progresses, the number of abnormal white blood cells increases rapidly. Symptoms may become more severe, including fatigue, weight loss, and easy bleeding or bruising.
- Blast crisis: This is the most advanced stage, characterized by a high number of immature white blood cells (blasts) in the blood and bone marrow. Symptoms can be severe and life-threatening, including fever, infections, and bleeding.
What happens if CML is not treated?
If chronic myeloid leukemia (CML) is not treated, the disease can progress through three phases: chronic, accelerated, and blast crisis. As the disease advances, symptoms become more severe, including fatigue, weight loss, easy bleeding, and infections. In the blast crisis stage, the disease can be life-threatening. Early diagnosis and treatment with tyrosine kinase inhibitors (TKIs) are crucial for managing CML and improving outcomes.
Does CML run in families?
Chronic myeloid leukemia (CML) is not typically inherited. While a small number of cases of chronic myeloid leukemia (CML) may be linked to inherited genetic factors, most cases of chronic myeloid leukemia (CML) occur spontaneously. However, certain genetic mutations, such as the Philadelphia chromosome, are common in chronic myeloid leukemia (CML) and can be detected through genetic testing.
Can CML spread to other organs?
Yes, chronic myeloid leukemia (CML) can spread to other organs. As the disease progresses, abnormal white blood cells can accumulate in various organs, including the liver, spleen, and lymph nodes. This can lead to organ enlargement, pain, and dysfunction. However, with effective treatment, the spread of chronic myeloid leukemia (CML) can be controlled, and complications can be minimized.
Disclaimer: This article is intended for informational purposes only and is specifically targeted towards medical students. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Alaggio R, Amador C, Anagnostopoulos I, Attygalle AD, Araujo IBO, Berti E, Bhagat G, Borges AM, Boyer D, Calaminici M, Chadburn A, Chan JKC, Cheuk W, Chng WJ, Choi JK, Chuang SS, Coupland SE, Czader M, Dave SS, de Jong D, Du MQ, Elenitoba-Johnson KS, Ferry J, Geyer J, Gratzinger D, Guitart J, Gujral S, Harris M, Harrison CJ, Hartmann S, Hochhaus A, Jansen PM, Karube K, Kempf W, Khoury J, Kimura H, Klapper W, Kovach AE, Kumar S, Lazar AJ, Lazzi S, Leoncini L, Leung N, Leventaki V, Li XQ, Lim MS, Liu WP, Louissaint A Jr, Marcogliese A, Medeiros LJ, Michal M, Miranda RN, Mitteldorf C, Montes-Moreno S, Morice W, Nardi V, Naresh KN, Natkunam Y, Ng SB, Oschlies I, Ott G, Parrens M, Pulitzer M, Rajkumar SV, Rawstron AC, Rech K, Rosenwald A, Said J, Sarkozy C, Sayed S, Saygin C, Schuh A, Sewell W, Siebert R, Sohani AR, Tooze R, Traverse-Glehen A, Vega F, Vergier B, Wechalekar AD, Wood B, Xerri L, Xiao W. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia. 2022 Jul;36(7):1720-1748. doi: 10.1038/s41375-022-01620-2. Epub 2022 Jun 22. Erratum in: Leukemia. 2023 Sep;37(9):1944-1951. PMID: 35732829; PMCID: PMC9214472.
- Carr JH. Clinical Hematology Atlas 6th Edition (Elsevier).
- Jabbour E, Kantarjian H. Chronic myeloid leukemia: 2022 update on diagnosis, therapy, and monitoring. Am J Hematol. 2022 Sep;97(9):1236-1256. doi: 10.1002/ajh.26642. Epub 2022 Jul 6. PMID: 35751859.
- Jabbour EJ, Sasaki K, Haddad FG, Issa GC, Garcia-Manero G, Kadia TM, Jain N, Yilmaz M, DiNardo CD, Patel KP, Kanagal-Shamanna R, Champlin R, Khouri IF, Dellasala S, Pierce SA, Kantarjian H. The outcomes of patients with chronic myeloid leukemia treated with third-line BCR::ABL1 tyrosine kinase inhibitors. Am J Hematol. 2023 Apr;98(4):658-665. doi: 10.1002/ajh.26852. Epub 2023 Jan 31. PMID: 36683287.
- https://www.lls.org/booklet/chronic-myeloid-leukemia
- Javidi-Sharifi N, Hobbs G. Future Directions in Chronic Phase CML Treatment. Curr Hematol Malig Rep. 2021 Dec;16(6):500-508. doi: 10.1007/s11899-021-00658-w. Epub 2021 Oct 14. PMID: 34648120.
- Eden RE, Coviello JM. Chronic Myelogenous Leukemia. [Updated 2023 Jan 16]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK531459/
- Jabbour, E., & Kantarjian, H. (2024). Chronic myeloid leukemia: 2025 update on diagnosis, therapy, and monitoring. American journal of hematology, 99(11), 2191–2212. https://doi.org/10.1002/ajh.27443
- Jabbour E, Kantarjian H. Chronic Myeloid Leukemia: A Review. JAMA. 2025;333(18):1618–1629. https://doi.org/10.1001/jama.2025.0220