Key Takeaways
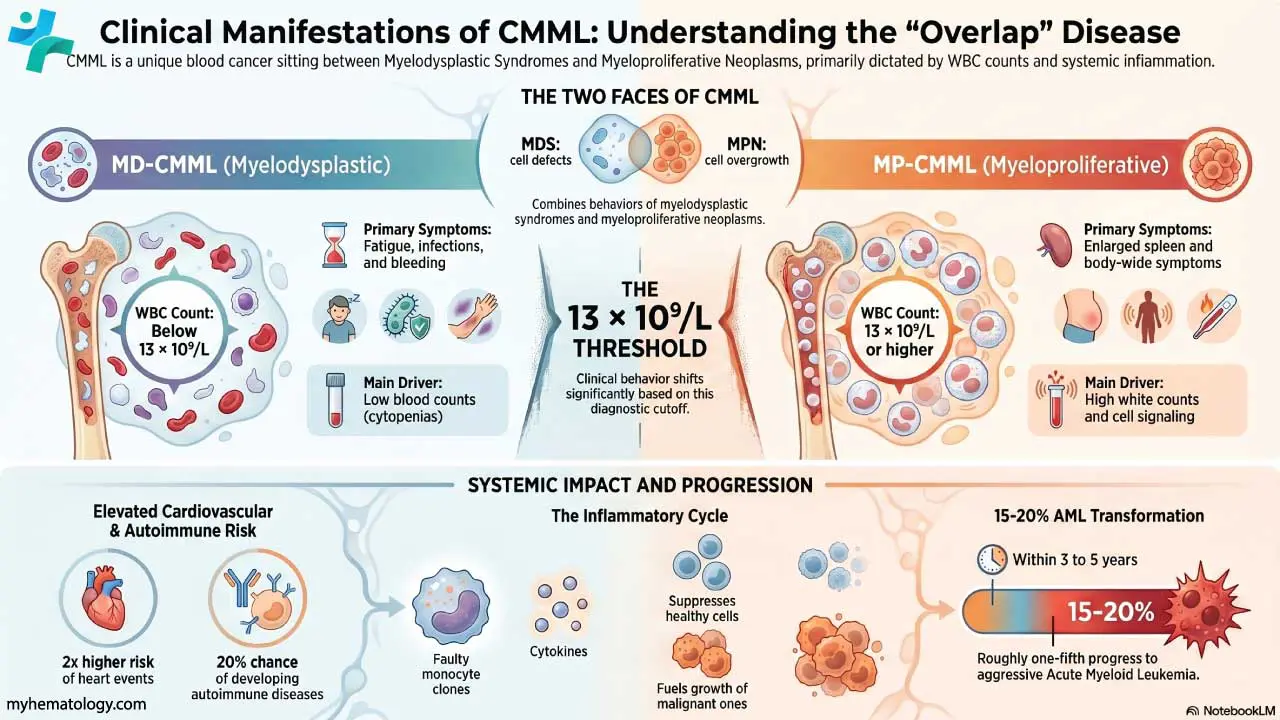
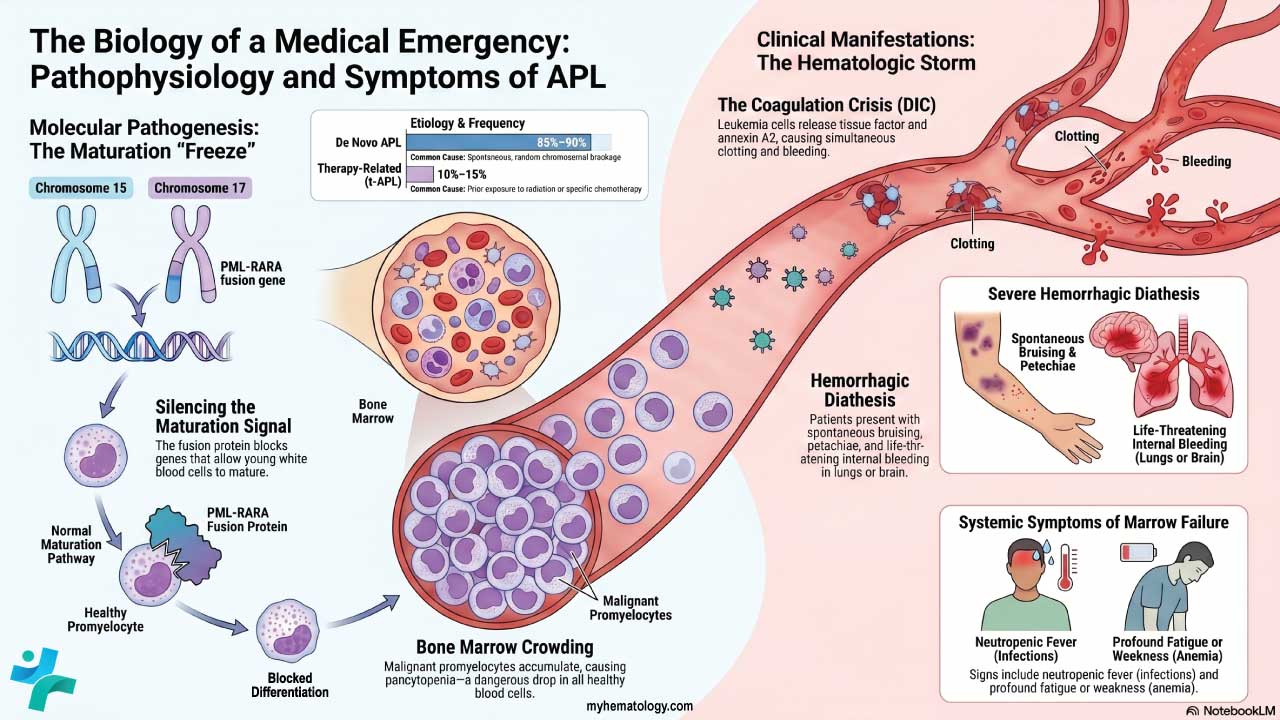
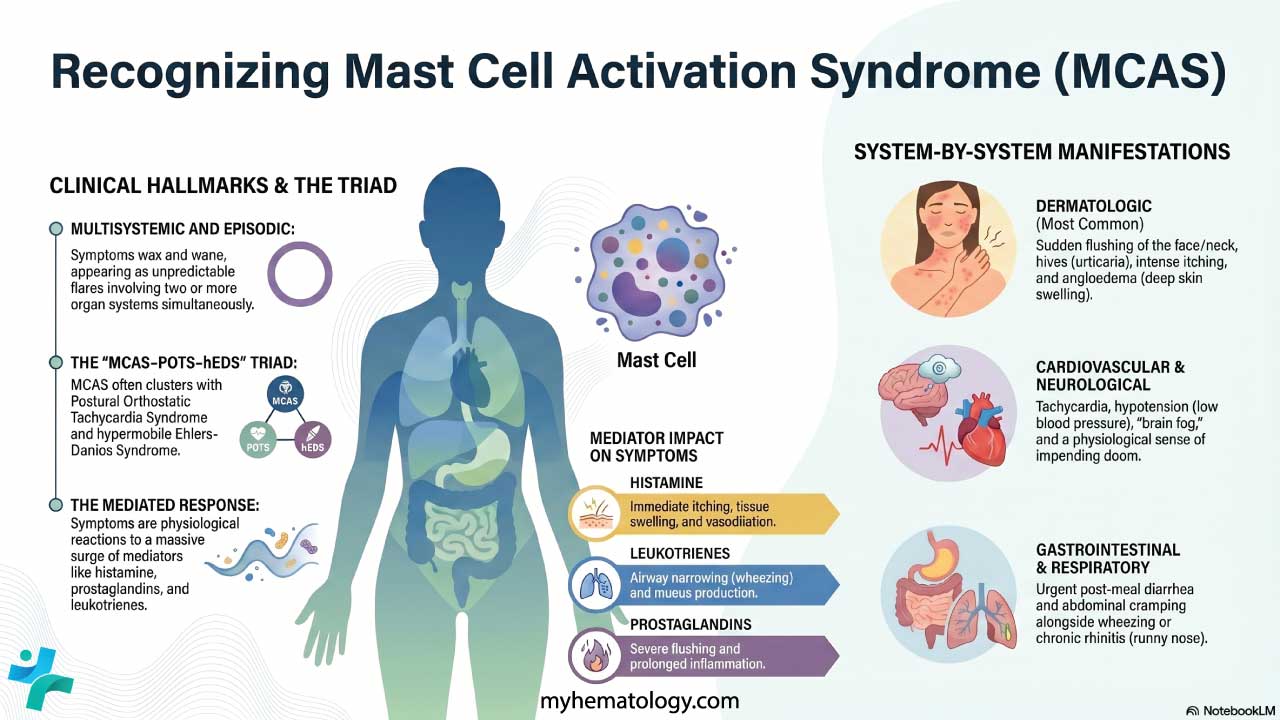
T-cell acute lymphoblastic leukemia (T-ALL) is an aggressive blood cancer caused by uncontrolled growth of immature T-cells (lymphoblasts) that fail to mature in the thymus [1,2].
- Epidemiology: T-ALL accounts for about 12–15% of pediatric ALL and 20–25% of adult ALL, with a strong male predominance (roughly 3:1) and a peak in adolescence and young adulthood [2,3].
- Causes ▾: More than 60% of cases carry activating mutations in the NOTCH1 gene; other key drivers include TAL1, TLX1/3, LMO1/2, and deletions of CDKN2A/B [4].
- Clinical features ▾: A chest (mediastinal) mass arising from the thymus is the clinical hallmark and is present in up to 75% of patients, sometimes causing superior vena cava syndrome.
- Diagnosis ▾: Diagnosis requires ≥20% lymphoblasts in marrow plus flow cytometry showing cytoplasmic CD3 with CD7, CD5, and CD2 [2].
- Treatment ▾: Modern frontline treatment combines multi-phase chemotherapy with nelarabine and pegaspargase [5,6]. For relapsed or refractory disease, next-generation targeted approaches such as allogeneic (off-the-shelf) CRISPR-engineered CD7-directed CAR-T cell therapies are reshaping clinical outcomes [10,11].
*Click ▾ for more information
What Is Acute Lymphoblastic Leukemia?
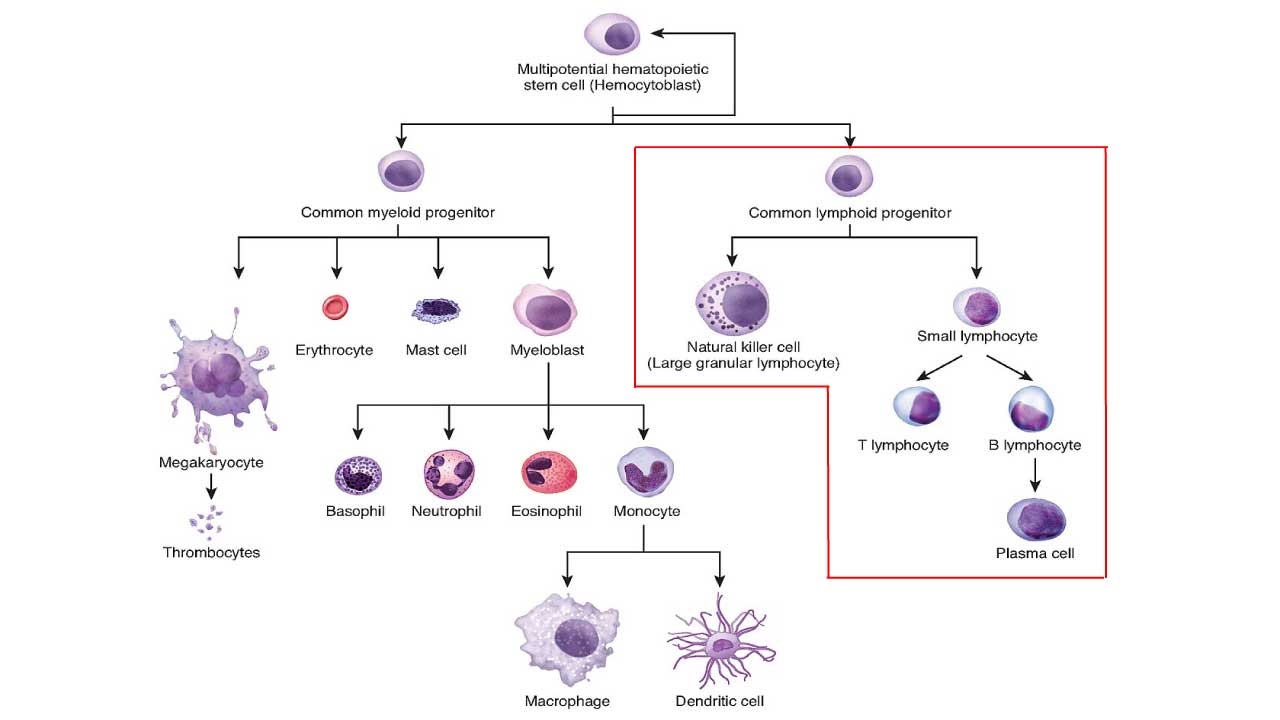
Acute lymphoblastic leukemia (ALL) is a cancer that starts in the bone marrow, the spongy tissue inside bones where blood cells are made. In ALL, the marrow churns out too many immature lymphocytes, called lymphoblasts. These blasts cannot fight infection, and they crowd out normal red cells, platelets, and healthy white cells. ALL is divided into two main types: B-cell ALL (B-ALL) and T-cell ALL (T-ALL), based on which lymphocyte lineage the blasts come from.
ALL is the most common cancer in children, with a peak between ages 2 and 5, and a second smaller peak after age 65 [2].
T-Cell Acute Lymphoblastic Leukemia
T-cell acute lymphoblastic leukemia begins in T-cell precursors that are still maturing in the thymus, the small organ behind the breastbone. When mutations interrupt that maturation, the blasts accumulate in the thymus and bone marrow, then spill into the blood and into organs such as the liver, spleen, lymph nodes, and central nervous system [2,4]. Compared with B-ALL, T-ALL is more likely to involve the chest, the brain, and the testes, and it tends to present with very high white blood cell counts.
Epidemiology
T-ALL has a distinct demographic fingerprint:
- Pediatric cases: roughly 12–15% of childhood ALL [2].
- Adult cases: roughly 20–25% of adult ALL [2,3].
- Age: peak in adolescence and young adulthood, with a median age around 15 years; a smaller bump occurs around age 30. Unlike B-ALL, T-ALL incidence does not climb sharply in older adults [2].
- Sex: males are about three times more likely to be affected than females. The reason is not fully understood but may involve X-chromosome tumor suppressor genes [2].
- Ancestry: higher relative frequency in people of African and Hispanic descent and lower frequency in East and Southeast Asian populations [2].
- ETP-ALL: the early T-cell precursor subtype represents about 15% of pediatric and up to 35% of adult T-ALL cases [2,3].
Survival has improved dramatically. In high-income settings, long-term survival now exceeds 85% in children, and recent adult regimens combining Hyper-CVAD with nelarabine, pegaspargase, and venetoclax have reported 2-year overall survival around 88% [5,6].
Causes and Risk Factors
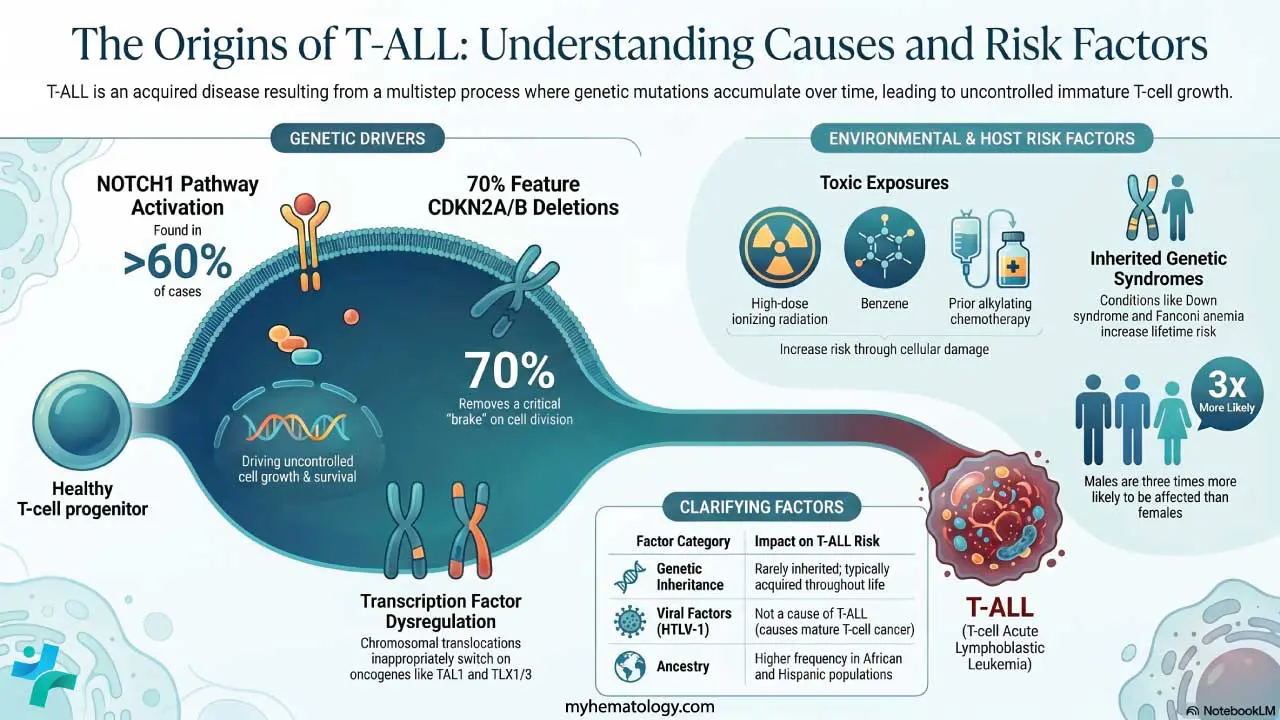
T-ALL is almost always acquired — caused by mutations that arise during life rather than inherited. It is best understood as a multistep process in which several genetic hits accumulate inside developing T-cells.
Genetic drivers
- NOTCH1 pathway activation. Found in over 60% of cases. Mutations in NOTCH1, or its regulator FBXW7, jam the receptor "on" and drive uncontrolled growth and survival [4,7].
- Transcription factor dysregulation. Chromosomal translocations move oncogenes such as TAL1/SCL, TLX1, TLX3, and LMO1/2 next to T-cell receptor (TCR) enhancers, switching them on inappropriately [4].
- Cell cycle release. Deletion of CDKN2A/B on chromosome 9p21 occurs in about 70% of cases, removing a key brake on cell division [4].
Environmental and host factors
- Ionizing radiation at high doses, including atomic bomb exposure and prior therapeutic radiation, raises leukemia risk.
- Previous chemotherapy with alkylating agents can cause therapy-related leukemia.
- Benzene exposure from heavy industrial solvents or chronic cigarette smoke is a known marrow toxin.
- Inherited syndromes that impair DNA repair or immunity including ataxia-telangiectasia, Li-Fraumeni syndrome, Down syndrome, Fanconi anemia, and Bloom syndrome increase the lifetime risk of T-ALL.
Does HTLV-1 Cause T-ALL?
HTLV-1 (human T-cell leukemia virus-1) causes adult T-cell leukemia/lymphoma, which is a mature T-cell cancer, not T-ALL. Viral causes are not established for T-ALL itself.
Clinical Features of T-ALL
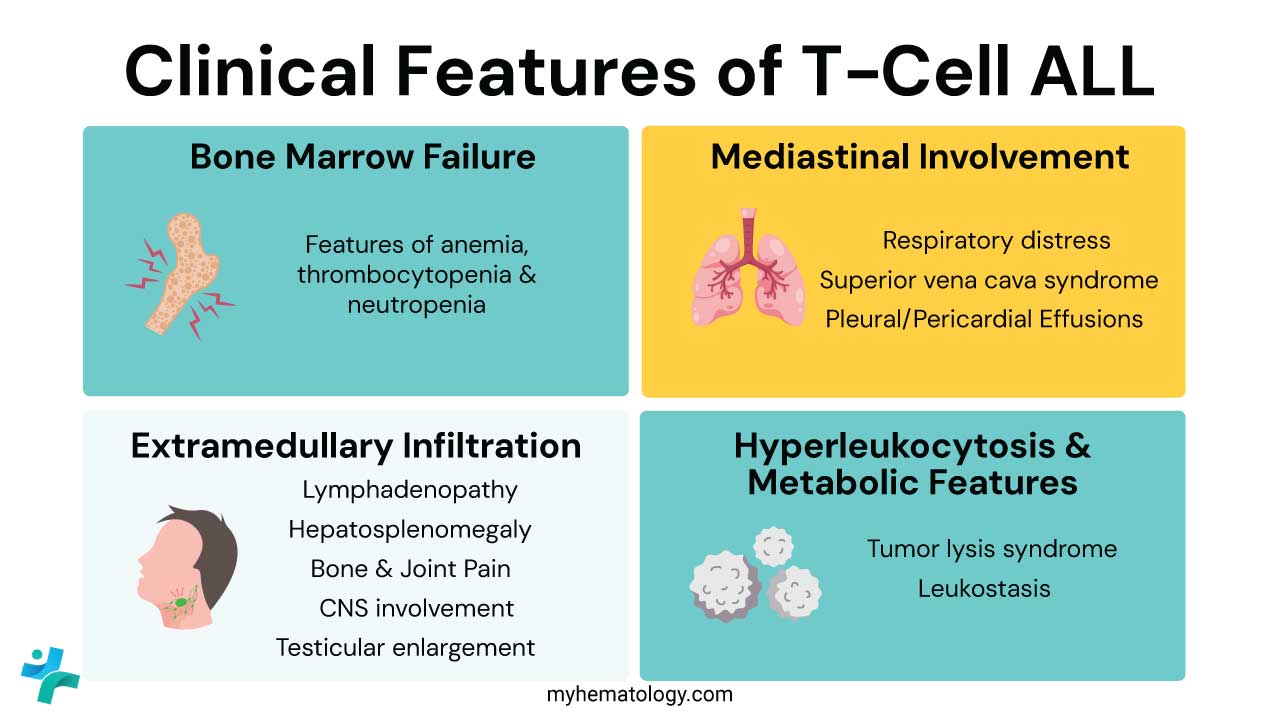
The signs of T-ALL fall into three groups: bone marrow failure, chest involvement, and spread to other organs.
Bone marrow failure
When blasts replace normal marrow, three deficiencies follow, classically called the bone marrow failure triad:
- Anemia: fatigue, breathlessness, pallor.
- Neutropenia: recurrent fevers and serious infections.
- Thrombocytopenia: bleeding gums, nosebleeds, petechiae, and easy bruising.
Mediastinal (chest) mass — the T-ALL hallmark
Up to three-quarters of patients have a large, fast-growing mass in the front of the chest, arising from the thymus [2]. Depending on what the mass presses on, it can cause:
- Breathing problems. Persistent cough, wheezing, or shortness of breath. It can be misdiagnosed as asthma in a teenager.
- Superior vena cava (SVC) syndrome. A medical emergency. Facial swelling, distended neck veins, and a flushed or bluish upper body. Symptoms typically worsen when the patient lies flat.
- Pleural or pericardial effusions. Fluid around the lungs or heart, worsening breathing and circulation.
Spread beyond the marrow
T-ALL frequently colonizes other tissues:
- Organomegaly. Enlarged liver (hepatomegaly) and spleen (splenomegaly), often with abdominal fullness.
- Lymphadenopathy. Painless, firm swelling of lymph nodes in the neck, armpits, and groin.
- Central nervous system (CNS) involvement. More common than in B-ALL. Headaches, morning vomiting, or cranial nerve palsies (such as facial drooping or double vision) [2].
- Testicular involvement. Painless, one-sided testicular enlargement; uncommon at diagnosis but a known sanctuary site.
- Bone pain. From leukemic expansion within the marrow cavity, sometimes causing children to refuse to walk.
Hyperleukocytosis and metabolic problems
Many T-ALL patients arrive with white cell counts above 100,000/µL [2], which carries two specific risks:
- Tumor lysis syndrome (TLS). Rapid cell turnover releases potassium, phosphate, and uric acid into the blood, threatening the kidneys and heart — sometimes before chemotherapy even begins.
- Leukostasis. Extreme white cell counts can sludge in small vessels, causing hypoxia, visual changes, or confusion.
Clinical red flags
- A teenager with new "asthma" that doesn't respond to inhalers (mediastinal mass).
- Facial fullness or neck vein distention mistaken for an allergic reaction (SVC syndrome).
- A white cell count above 100,000/µL.
- Severe breathlessness specifically when lying flat (orthopnea).
- Rapidly enlarging liver, spleen, or lymph nodes.
Laboratory Investigations and Diagnosis
Diagnosis of T-cell Acute Lymphoblastic Leukemia (T-cell ALL) requires a multi-modal approach.
Initial Blood Work (The Screening Phase)
- Complete blood count (CBC): typically shows marked leukocytosis, normocytic anemia, and thrombocytopenia.
- Peripheral blood smear: lymphoblasts in T-ALL often have a folded ("convoluted") nucleus, dense chromatin, and scant cytoplasm. Auer rods, which are seen in acute myeloid leukemia (AML), are never present.
- Metabolic panel: screens for tumor lysis syndrome, looking for high uric acid, high phosphate, high potassium, and elevated lactate dehydrogenase (LDH).
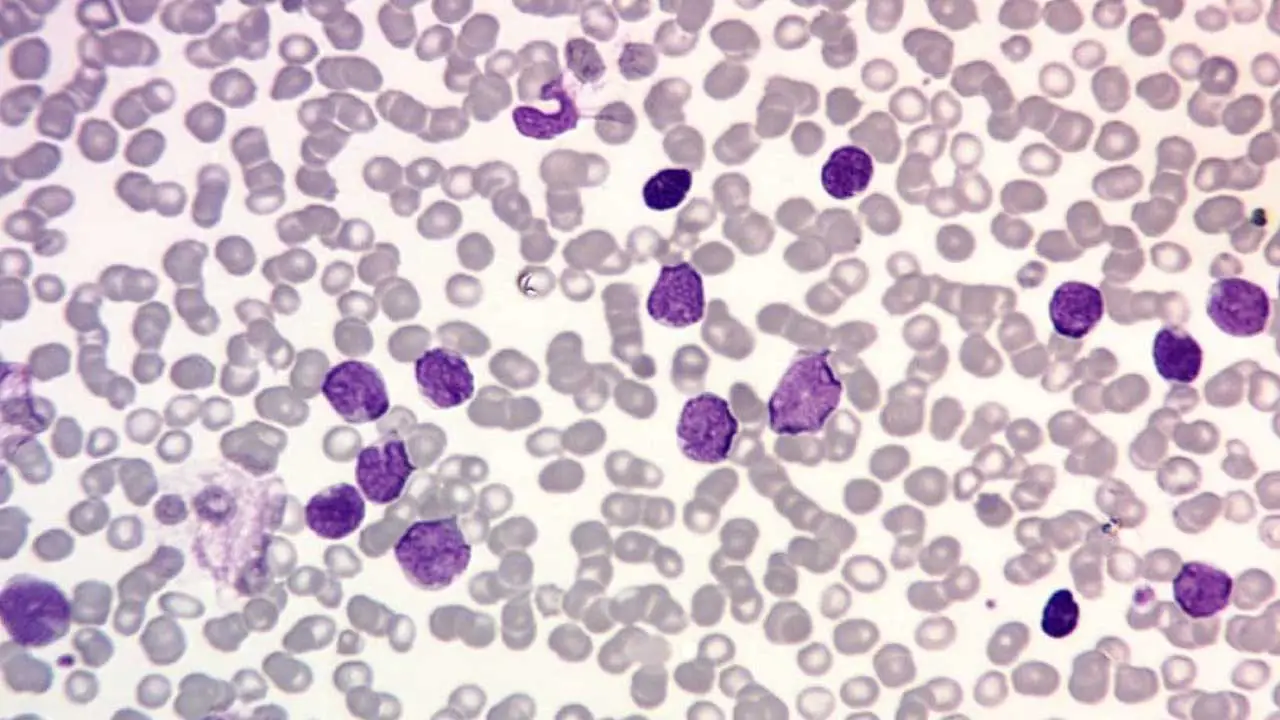
Bone Marrow Aspiration and Biopsy
This is the diagnostic gold standard. T-ALL is confirmed when ≥20% of marrow cells are lymphoblasts. On classical staining, blasts are periodic acid-Schiff (PAS) positive (chunky pattern) and myeloperoxidase (MPO) negative.
Flow Cytometry
Flow cytometry separates T-ALL from B-ALL and AML. It uses antibodies to detect proteins on or inside the cell.
- Most specific T-lineage marker: cytoplasmic CD3 (cCD3).
- Pan-T markers: CD7 (usually first to appear), CD5, CD2.
- Maturation stage markers: CD1a, CD4, CD8 — used to subclassify as pro-T, pre-T, cortical, or mature.
- ETP-ALL signature: CD7 positive; CD5 weak or negative on under 75% of blasts; CD1a and CD8 negative; with positivity for stem/myeloid markers such as CD34, CD13, CD33, or CD117 [2].
Cytogenetics and molecular profiling
- Karyotype and FISH: look for translocations involving the T-cell receptor loci at 14q11 (TCR α/δ) and 7q34 (TCR β).
- Next-generation sequencing (NGS): detects NOTCH1/FBXW7 mutations (over 60% of cases, often associated with better initial response), TAL1, TLX1, TLX3 rearrangements, and JAK/STAT pathway mutations (typical of ETP-ALL) [4,7]. Ultra-sensitive NGS is expanding across clinical centers for Minimal Residual Disease (MRD) monitoring, capable of tracking T-cell receptor (TCR) gene rearrangements down to a deep sensitivity of 1×106 (1 leukemic cell per million), offering vastly superior relapse prognostication over standard flow cytometry [12,13].
Assessing spread
- Lumbar puncture: mandatory at diagnosis to check spinal fluid for leukemic cells, even if the patient has no neurological symptoms.
- Imaging: chest X-ray and CT to assess any mediastinal mass and airway compression; abdominal ultrasound or CT to document organomegaly.
Diagnostic Summary Table
Treatment and Management of T-Cell ALL
Management of T-cell acute lymphoblastic leukemia balances intensive chemotherapy with newer targeted drugs. Treatment runs over 2 to 3 years and is divided into four phases. Before any of that begins, oncologic emergencies must be controlled.
First 24 to 48 hours: managing emergencies
- Tumor lysis syndrome prophylaxis. Aggressive IV fluids and rasburicase (recombinant urate oxidase) prevent acute kidney injury.
- Mediastinal mass and SVC syndrome. High-dose dexamethasone shrinks the mass quickly. Patients are kept upright. General anesthesia is avoided when possible because of the risk of total airway collapse.
Induction: getting to remission
The goal is complete remission (CR) with under 5% blasts in the marrow. Standard regimens include Hyper-CVAD or pediatric-inspired BFM-style protocols, built around vincristine, dexamethasone, an anthracycline (doxorubicin or daunorubicin), and pegaspargase [3,5]. Nelarabine, a purine analog uniquely toxic to T-lymphoblasts, is now incorporated into frontline therapy for high-risk T-ALL based on the COG AALL0434 trial [5].
Consolidation and CNS prophylaxis
- Intensification. High-dose methotrexate and cytarabine penetrate sanctuary sites such as the central nervous system and the testes.
- CNS protection. Every T-ALL patient receives intrathecal chemotherapy — methotrexate or cytarabine injected into the spinal fluid. Cranial radiation is now reserved for patients with proven CNS disease at diagnosis [2,3].
- MRD monitoring. Minimal residual disease, measured by flow cytometry or NGS, is the single strongest predictor of relapse. Patients who remain MRD-positive after induction are fast-tracked toward stem cell transplant [3].
Maintenance and stem cell transplant
- Maintenance. Two to three years of low-intensity oral therapy, mainly daily 6-mercaptopurine and weekly methotrexate, to mop up residual disease.
- Allogeneic stem cell transplant. The definitive option for high-risk patients: ETP-ALL with persistent MRD, induction failure, or relapsed disease in second remission [3].
Emerging and targeted therapies
The field is moving fast. Several approaches have changed the conversation in the past five years:
- Venetoclax (BCL-2 inhibitor). Adding venetoclax to nelarabine and pegaspargase within Hyper-CVAD has produced 2-year overall survival around 88% in adult T-ALL trials [6]. It is being actively studied in ETP-ALL, which is historically chemo-resistant [3].
- Allogeneic CD7-directed CAR-T cell therapy: CD7 is present on virtually all T-ALL blasts, but engineered T-cells naturally express CD7 too, leading to self-destruction (fratricide). In early 2026, the FDA granted Breakthrough Therapy Designation to soficabtagene geleucel (sofi-cel; formerly WU-CART-007), a first-in-class, allogeneic (“off-the-shelf”) anti-CD7 CAR-T cell therapy manufactured from healthy donor cells [10]. In clinical trial results, sofi-cel demonstrated an overall response rate of 91%, with 72.7% of relapsed/refractory T-ALL patients achieving complete remission at the recommended phase 2 dose, providing an immediate, pre-manufactured bridge to a curative stem cell transplant without typical manufacturing delays [11].
- Daratumumab (anti-CD38) and bortezomib are being added to pediatric and adolescent regimens for high-risk disease [7].
- JAK inhibitors such as ruxolitinib are under study for JAK/STAT-mutated ETP-ALL [7].
What Caregivers Can Expect
Treatment usually starts in the hospital. The first weeks are the most intense with close monitoring for tumor lysis syndrome, a risk of serious infections because of low neutrophil counts, and supportive care for nausea, mouth sores, and fatigue. Hair loss is common but temporary. Once the patient enters maintenance, most care moves to the outpatient clinic. Long-term follow-up watches for late effects: heart effects from anthracyclines, fertility issues, and neurocognitive changes from CNS-directed therapy. Whenever possible, fertility preservation should be discussed before induction begins, especially for adolescents and young adults.
Summary of B-cell ALL vs T-cell ALL
Frequently Asked Questions (FAQs)
Is T-cell acute lymphoblastic leukemia (T-ALL) leukemia curable?
Yes, often. With modern protocols, long-term survival exceeds 85% in children and reaches roughly 60–70% in adolescents and young adults treated in well-resourced centers [2,3]. Recent adult regimens combining Hyper-CVAD with nelarabine, pegaspargase, and venetoclax have reported 2-year overall survival close to 88% [6]. Outcomes are still poorer for ETP-ALL and relapsed disease, which is why MRD monitoring and access to transplant are so important.
Why does T-ALL often cause breathing problems?
T-ALL begins in the thymus, which sits behind the breastbone. As blasts multiply, they form a chest mass that can press on the windpipe or on the superior vena cava. This causes coughing, wheezing, breathlessness, or facial swelling. Up to three-quarters of patients have this feature at diagnosis [2].
What makes ETP-ALL different from standard T-ALL?
Early T-cell precursor (ETP) ALL arises from very immature cells that still carry stem-cell or myeloid features. It tends to resist standard chemotherapy, so most patients are considered for an allogeneic stem cell transplant in first remission [3]. Newer combinations with venetoclax look promising but are still under study.
Can T-ALL spread to the brain?
Yes. T-ALL has a higher tendency to involve the central nervous system than B-ALL. Every T-ALL treatment plan therefore includes intrathecal chemotherapy, given by lumbar puncture, to treat or prevent CNS disease [2].
Is the Philadelphia chromosome found in T-ALL?
Almost never. The Philadelphia chromosome (t(9;22)) is a hallmark of a subset of B-ALL. T-ALL is driven by different genetic changes — most often NOTCH1 mutations and rearrangements involving TAL1, TLX1, or TLX3 [4].
What is CAR-T therapy and is it used in T-ALL?
CAR-T therapy re-engineers immune cells to recognize and destroy cancer. While highly successful in B-ALL, engineering CAR-T for T-ALL historically faced a major barrier called fratricide, where the therapeutic T-cells attack one another because they both share the CD7 protein marker [11]. In January 2026, the FDA granted Breakthrough Therapy Designation to an allogeneic (“off-the-shelf”) CRISPR-engineered therapy called soficabtagene geleucel (sofi-cel) [10].Because it is pre-manufactured from healthy donor cells rather than a patient's own heavily pretreated cells, it avoids fatal manufacturing delays. By utilizing gene editing to knock out CD7 and TRAC genes, it prevents fratricide and graft-versus-host disease, successfully clearing the disease and providing an effective bridge to stem cell transplantation in over 70% of heavily pretreated patients [11].
Glossary of Related Medical Terms
- Lymphoblast: Immature white blood cell that has not matured into a working lymphocyte.
- Thymus: Small organ behind the breastbone where T-cells normally mature.
- Mediastinal mass: Tumor in the central chest, usually from the thymus in T-ALL.
- Superior vena cava (SVC) syndrome: Compression of the main vein returning blood from the upper body. A medical emergency.
- Tumor lysis syndrome (TLS): Dangerous release of cell contents into the blood when cancer cells die rapidly.
- Cytoplasmic CD3 (cCD3): Protein inside developing T-cells; the most specific marker of T-cell lineage on flow cytometry.
- Flow cytometry: Lab test using antibodies and lasers to identify proteins on or inside cells.
- Early T-cell precursor (ETP) ALL: High-risk T-ALL subtype with stem-cell-like features.
- NOTCH1 mutation: "Always-on" change found in over 60% of T-ALL cases.
- Minimal residual disease (MRD): Tiny amounts of leukemia detectable only by sensitive lab tests; the strongest predictor of relapse.
- Intrathecal chemotherapy: Chemo injected into the spinal fluid through a lumbar puncture.
- Nelarabine: Chemo drug uniquely toxic to T-lymphoblasts.
- Pegaspargase: Long-acting form of asparaginase, an enzyme that starves blasts of asparagine.
- CAR-T cell therapy: Re-engineering a patient's T-cells to attack cancer.
- Fratricide: When CAR-T cells share a target with the cancer and attack each other; a key obstacle in T-ALL CAR-T design.
- Allogeneic stem cell transplant: Transplant of donor stem cells to replace diseased marrow..
Disclaimer: This article is intended for educational and informational purposes only. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Alaggio, R., Amador, C., Anagnostopoulos, I., Attygalle, A. D., Araujo, I. B. O., Berti, E., Bhagat, G., Borges, A. M., Boyer, D., Calaminici, M., Chadburn, A., Chan, J. K. C., Cheuk, W., Chng, W. J., Choi, J. K., Chuang, S. S., Coupland, S. E., Czader, M., Dave, S. S., de Jong, D., … Xiao, W. (2022). The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia, 36(7), 1720–1748. https://doi.org/10.1038/s41375-022-01620-2
- Raetz, E. A., & Teachey, D. T. (2016). T-cell acute lymphoblastic leukemia. Hematology. American Society of Hematology. Education Program, 2016(1), 580–588. https://doi.org/10.1182/asheducation-2016.1.580
- Kantarjian, H., & Jabbour, E. (2025). Adult Acute Lymphoblastic Leukemia: 2025 Update on Diagnosis, Therapy, and Monitoring. American journal of hematology, 100(7), 1205–1231. https://doi.org/10.1002/ajh.27708
- Fattizzo B, Rosa J, Giannotta JA, Baldini L and Fracchiolla NS (2020) The Physiopathology of T- Cell Acute Lymphoblastic Leukemia: Focus on Molecular Aspects. Front. Oncol. 10:273. https://doi.org/10.3389/fonc.2020.00273
- Dunsmore, K. P., Winter, S. S., Devidas, M., Wood, B. L., Esiashvili, N., Chen, Z., Eisenberg, N., Briegel, N., Hayashi, R. J., Gastier-Foster, J. M., Carroll, A. J., Heerema, N. A., Asselin, B. L., Rabin, K. R., Zweidler-Mckay, P. A., Raetz, E. A., Loh, M. L., Schultz, K. R., Winick, N. J., Carroll, W. L., … Hunger, S. P. (2020). Children's Oncology Group AALL0434: A Phase III Randomized Clinical Trial Testing Nelarabine in Newly Diagnosed T-Cell Acute Lymphoblastic Leukemia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology, 38(28), 3282–3293. https://doi.org/10.1200/JCO.20.00256
- Senapati J, Kantarjian HM, Jabbour E, et al. Nelarabine (NEL), Pegylated Asparginase (PEG) and Venetoclax (VEN) Incorporated to HCVAD Chemotherapy in the Frontline Treatment of Adult Patients with T-Cell Acute Lymphoblastic Leukemia/Lymphoblastic Lymphoma (T-ALL/T-LBL). Blood. 2023;142(Suppl 1):963. https://doi.org/10.1182/blood-2023-179562
- Caracciolo, D., Mancuso, A., Polerà, N., Froio, C., D'Aquino, G., Riillo, C., Tagliaferri, P., & Tassone, P. (2023). The emerging scenario of immunotherapy for T-cell Acute Lymphoblastic Leukemia: advances, challenges and future perspectives. Experimental hematology & oncology, 12(1), 5. https://doi.org/10.1186/s40164-022-00368-w
- Lu, P., Liu, Y., Yang, J., Zhang, X., Yang, X., Wang, H., Wang, L., Wang, Q., Jin, D., Li, J., & Huang, X. (2022). Naturally selected CD7 CAR-T therapy without genetic manipulations for T-ALL/LBL: first-in-human phase 1 clinical trial. Blood, 140(4), 321–334. https://doi.org/10.1182/blood.2021014498
- Hefazi, M., & Litzow, M. R. (2018). Recent Advances in the Biology and Treatment of T Cell Acute Lymphoblastic Leukemia. Current hematologic malignancy reports, 13(4), 265–274. https://doi.org/10.1007/s11899-018-0455-9
- Cure Today. FDA Grants Breakthrough Designation to Sofi-cel for Relapsed/Refractory T-Cell ALL/LBL. Cure Today. January 21, 2026.
- American Journal of Managed Care (AJMC) News. Off-the-Shelf CAR T-Cell Therapy Granted Breakthrough Therapy Designation for Aggressive T-Cell Cancers. AJMC Media. March 26, 2026.
- Short, N. J., Kantarjian, H., Ravandi, F., Konopleva, M., Jain, N., Kanagal-Shamanna, R., Patel, K. P., Macaron, W., Kadia, T. M., Wang, S., Jorgensen, J. L., Khoury, J. D., Yilmaz, M., Kebriaei, P., Takahashi, K., Garcia-Manero, G., Daver, N., Post, S. M., Huang, X., Kornblau, S. M., … Jabbour, E. (2022). High-sensitivity next-generation sequencing MRD assessment in ALL identifies patients at very low risk of relapse. Blood advances, 6(13), 4006–4014. https://doi.org/10.1182/bloodadvances.2022007378
- Muffly, L., Liang, E. C., Dolan, J. G., & Pulsipher, M. A. (2024). How I use next-generation sequencing-MRD to plan approach and prevent relapse after HCT for children and adults with ALL. Blood, 144(3), 253–261. https://doi.org/10.1182/blood.2023023699