Key Takeaways
Acute promyelocytic leukemia (APL) is a subtype of acute myeloid leukemia driven by the t(15;17) translocation, which creates the PML-RARA fusion gene and blocks white blood cells from maturing.
- Symptoms ▾: Patients typically present with the acute consequences of bone marrow failure, most dangerously a life-threatening bleeding disorder (DIC), alongside severe infections and profound fatigue.
- Investigations ▾: Diagnosis requires a parallel approach of immediate coagulation profiling and flow cytometry, followed by definitive genetic confirmation of the PML-RARA fusion via rapid FISH or PCR.
- Treatment ▾: Because APL is a true medical emergency, targeted therapy with all-trans retinoic acid (ATRA) and arsenic trioxide (ATO) must begin immediately upon clinical suspicion to resolve the coagulopathy and cure the leukemia.
*Click ▾ for more information
Why APL Deserves Your Full Attention
As you move from classroom pathology to clinical rotations, few conditions will be as intense or fast-moving as acute promyelocytic leukemia. Mastering it matters. APL is a genuine emergency that demands immediate recognition. Miss it, and a patient can bleed to death within hours. Manage it well, and you witness one of medicine's most striking turnarounds, from a near-certain killer to a curable disease.
This guide walks through APL the way the disease actually unfolds in the clinic: the paradox at its core, the biology that drives it, the first frantic 24 hours, the targeted drugs that cure it, and the complications you must anticipate.
The APL Paradox: Deadly Early, Curable Later
APL accounts for roughly 5% to 15% of acute myeloid leukemia cases, with about 600 to 800 new diagnoses each year in the United States [1]. It carries a striking contradiction.
In its first days, APL is lethal. The leukemia cells unleash a bleeding disorder so severe that patients can die before treatment takes hold. Here the numbers deserve a careful eye. Large clinical trials report early death rates of about 5% to 10%. But real-world, population-based studies, which include older and sicker patients that trials often exclude, report early death rates reaching up to 30% [8]. As a future clinician, remember the higher number. It reflects the patients you will actually meet.
Survive the first 30 days, though, and the outlook flips. Five-year overall survival climbs above 90% [2]. The lesson is simple and worth memorizing: the entire battle is won or lost early. The priority is to carry the patient through the emergency so the cure can do its work.
Pathophysiology
To read the clinical picture, start with the molecular fault. APL is driven by a balanced chromosomal translocation written as t(15;17). This swap fuses the promyelocytic leukemia (PML) gene on chromosome 15 with the retinoic acid receptor alpha (RARA) gene on chromosome 17.
The result is the PML-RARA fusion protein. Normally, retinoic acid switches on the genes that let young white blood cells mature. The fusion protein silences those genes instead. White blood cell development stalls at the promyelocyte stage, an early, immature step. The bone marrow then fills with these malignant promyelocytes. This crowding produces pancytopenia, a drop in all blood cell types, and sets the stage for severe complications.
Etiology: Spontaneous vs. Therapy-Related Disease
The t(15;17) chromosomal translocation develops via two distinct clinical pathways:
- De Novo APL: Comprising roughly 85% to 90% of all diagnoses, this form occurs spontaneously due to random, somatic chromosomal breakage and misrepair during myelopoiesis. It carries no identifiable lifestyle, environmental, or inherited genetic risk factors [4].
- Therapy-Related APL (t-APL): Accounting for 10% to 15% of cases, t-APL is a secondary malignancy directly linked to prior exposure to DNA topoisomerase II inhibitors (such as mitoxantrone, etoposide, doxorubicin, or epirubicin) or radiation therapy. This form is most commonly identified in patients with a history of treated breast cancer or lymphoma [10].
Why APL Bleeds: Tissue Factor and Annexin A2
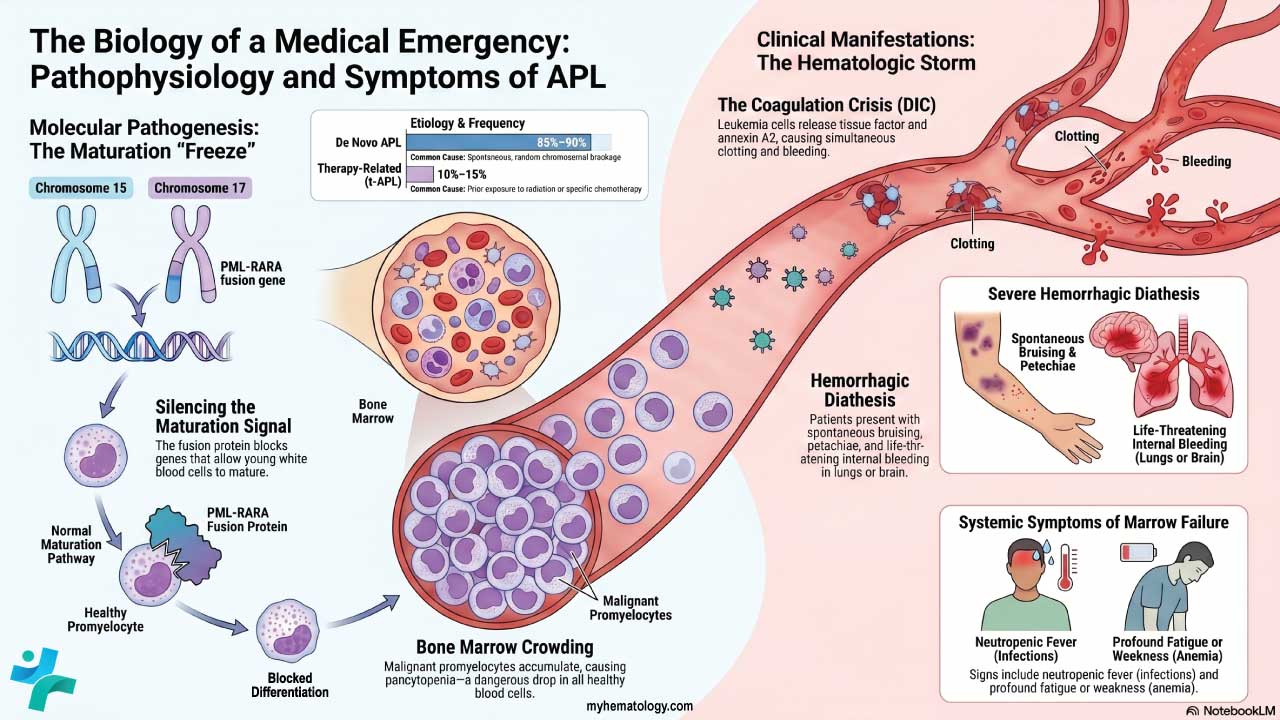
The flood of leukemic promyelocytes triggers a life-threatening coagulopathy, a breakdown in normal clotting. These abnormal cells are packed with granules rich in pro-clotting material. As the cells die or break apart, they spill tissue factor into the blood, which massively activates the clotting cascade. At the same time, the cells express high levels of annexin A2, a surface protein that ramps up the breakdown of clots (fibrinolysis) [3].
So clotting and clot breakdown surge together. This is disseminated intravascular coagulation, or DIC, a state where the blood both clots and bleeds out of control. Nearly 90% of patients bleed, and a substantial fraction suffer catastrophic hemorrhage into the brain or lungs [3]. This coagulopathy, not the leukemia itself, is what kills patients early.
Clinical Manifestations
As malignant promyelocytes accumulate rapidly and crowd out healthy hematopoiesis, patients experience an acute clinical onset driven by profound cytopenias and systemic coagulopathy:
Severe Hemorrhagic Diathesis: Extensive, spontaneous ecchymoses (bruising), petechiae, gingival bleeding, epistaxis, or life-threatening intracranial and pulmonary hemorrhages driven by the sudden onset of DIC [4].
Infectious Complications: Neutropenic fever, shaking chills, and severe localized bacterial or fungal infections secondary to the functional absence of mature white blood cells [2].
Anemic Symptoms: Acute onset of profound fatigue, generalized weakness, exertional dyspnea, and visible mucosal pallor resulting from a precipitous drop in red blood cells.
Clinical Red Flags: When to Suspect APL
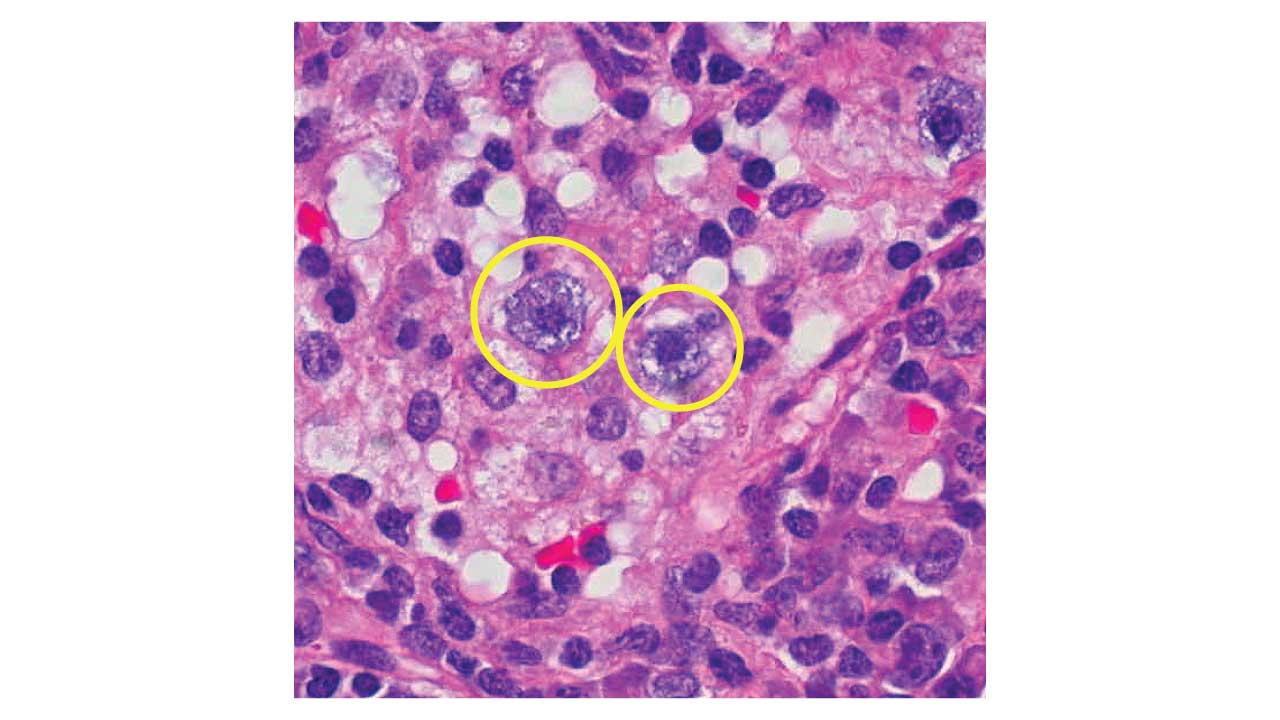
Keep your suspicion high in the emergency department. Watch for unexplained bruising, bleeding from the gums or other mucous membranes, and petechiae (tiny red skin spots). Pair these with a low platelet count and prolonged clotting times, the prothrombin time (PT) and activated partial thromboplastin time (aPTT). A peripheral blood smear seals the suspicion: atypical promyelocytes with heavy granules and Auer rods, needle-like inclusions that often stack into bundles in APL.
Investigations
Diagnosing APL requires a rapid, parallel diagnostic approach aimed at mapping the severity of the coagulopathy while simultaneously identifying the genetic driver:
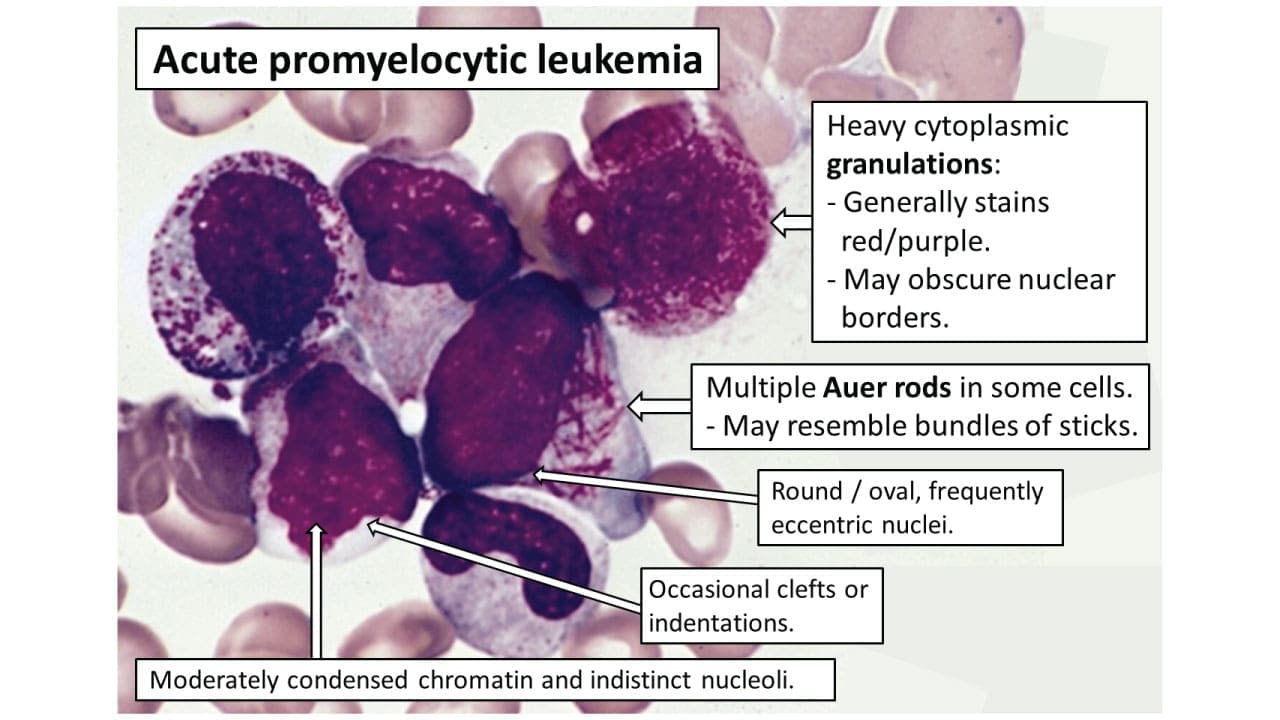
- Hematologic Testing: A complete blood count (CBC) typically reveals pancytopenia, though up to 30% of patients present with high-risk leukocytosis (WBC > 10 x 109/L). A peripheral blood smear provides the first presumptive morphological clue: atypical promyelocytes with folded or bilobed nuclei, heavy azurophilic granulation, and "faggot cells" containing stacked bundles of needle-like Auer rods [4].
- Coagulation Profiling: Immediate, serial screening must run every 6 to 12 hours to track the severity of the DIC. Typical findings include a prolonged prothrombin time (PT/INR), prolonged activated partial thromboplastin time (aPTT), massively elevated D-dimer, and a dangerously low fibrinogen level (frequently dropping below 150 mg/dL) [3].
- Flow Cytometry Immunophenotyping: This provides a rapid immunophenotypic profile within hours of sample collection. Leukemic promyelocytes show a highly distinctive expression pattern: they are strongly positive for CD33, CD13, CD117, and myeloperoxidase (MPO), but crucially negative or dim for CD34 and HLA-DR [4].
- Cytogenetic and Molecular Confirmation: The absolute gold standard involves rapid Fluorescence In Situ Hybridization (FISH) and quantitative reverse-transcription PCR (RT-PCR). These modalities definitively identify the pathognomonic t(15;17) translocation and PML-RARA fusion transcripts within 24 to 48 hours, confirming the diagnosis and establishing a baseline for long-term minimal residual disease (MRD) monitoring [4].
The First 24 Hours
Because APL is a true oncologic emergency, the first 24 hours decide survival. Bleeding is the enemy. Aggressive supportive care to reverse the coagulopathy must run in parallel with definitive treatment, not after it [4].
Here is what that looks like in practice:
- Treat first, confirm later. Give all-trans retinoic acid (ATRA) the moment APL is suspected. Do not wait for genetic or bone marrow results [4].
- Avoid invasive procedures. Hold off on central lines, lumbar punctures, and surgery until the coagulopathy is controlled [4].
- Transfuse aggressively. Clinicians commonly aim to keep fibrinogen above about 150 mg/dL with cryoprecipitate, and platelets above roughly 30 to 50 x 109/L with platelet transfusions [4].
- Monitor closely. Recheck PT, aPTT, fibrinogen, and D-dimer every 6 to 12 hours [4].
- Protect the heart and brain. Arsenic trioxide (ATO) prolongs the QTc interval, risking fatal Torsades de Pointes; mandate daily ECGs and aggressively maintain potassium above 4.0 mEq/L and magnesium above 2.0 mg/dL. Additionally, monitor for severe headaches and vision changes, which are signs of pseudotumor cerebri (idiopathic intracranial hypertension), a known toxicity of ATRA [4].
These targets are widely used, but treat them as guides rather than rigid rules; protocols vary.
Beyond Chemotherapy: The Targeted Cure with ATRA and Arsenic Trioxide
The treatment of APL is a landmark in precision oncology. Rather than relying on chemotherapy to blindly destroy fast-dividing cells, modern therapy persuades the cancer cells to grow up.
Both lead drugs attack the root cause. ATRA, given at pharmacological doses, overrides the fusion protein's block, letting the leukemic promyelocytes mature into normal neutrophils that then die off naturally. Arsenic trioxide (ATO) works alongside it, helping break down the PML-RARA fusion protein. Acting on different parts of the same faulty protein, the two drugs together clear the leukemia. ATRA plus ATO is now the standard of care for non-high-risk patients [5,6].
Targeted Therapy vs. Standard Chemotherapy
| Feature | ATRA + ATO | Standard Chemotherapy |
| Mechanism | Forces cells to mature and breaks down the fusion protein | Poisons rapidly dividing cells |
| Long-term survival | Superior in randomized trials [5,6] | Lower [5,6] |
| Complete remission | ~90%+ [5,6] | High, but more relapse over time |
| Main side effects | Differentiation syndrome, QTc prolongation, liver toxicity | Severe marrow suppression, hair loss, heart toxicity (anthracyclines) |
The long-term data are striking. In the Italian-German APL0406 trial, ATRA-ATO was at least as effective as, and on extended follow-up superior to, ATRA plus chemotherapy in non-high-risk APL [5,6].
Managing Complications
Targeted therapy is powerful, but it brings its own dangers. As huge numbers of leukemia cells mature at once, they release a storm of inflammatory signals and migrate into tissues. This is differentiation syndrome.
It affects roughly 25% to 30% of patients on ATRA [2]. Warning signs include unexplained fever, breathing difficulty, lung infiltrates, weight gain, and fluid around the lungs or heart. Untreated, it can progress to kidney failure and death. On top of this, patients are profoundly neutropenic during early induction, leaving them dangerously exposed to bacterial and fungal infection.
Steroids and the Cytokine Storm
Act fast. At the first sign of breathing trouble, weight gain, or unexplained fever in a patient on induction therapy, immediately start intravenous dexamethasone at 10 mg twice daily. ATRA and ATO are generally continued during mild to moderate differentiation syndrome and are only temporarily halted if the syndrome becomes severe, such as requiring mechanical ventilation or causing acute renal failure. Many modern protocols give preventive corticosteroids during the first weeks of induction to head off the cytokine storm entirely. Broad-spectrum preventive antibiotics are added to cover the high infection risk during the neutropenic window [4].
The Road to Remission and the Latest Evidence
The patient's journey is remarkable. On day zero, they face catastrophic bleeding. By day 30, with the coagulopathy and any differentiation syndrome controlled, the immediate threat to life recedes. By year five, most patients are considered cured.
After remission, monitoring does not stop. Doctors track the PML-RARA gene with sensitive PCR blood tests to catch any early sign of relapse, which is uncommon but treatable when found promptly.
High-Risk APL: What the APOLLO Trial Changed
High-risk APL, usually defined by a white blood cell count above 10 x 109/L at diagnosis, has long been treated with added anthracycline chemotherapy to rein in the rapidly multiplying cells. That picture is shifting. The phase III APOLLO trial compared an ATRA-ATO–based regimen with low-dose idarubicin against standard ATRA-chemotherapy in high-risk patients. The ATRA-ATO arm achieved significantly better 2-year event-free survival, about 88% versus 70%, along with fewer relapses and less toxicity [7].
Furthermore, another highly effective, chemotherapy-free standard for high-risk APL combines ATRA, ATO, and the targeted CD33 antibody-drug conjugate gemtuzumab ozogamicin (GO). Because high-risk patients also face increased risk of central nervous system (CNS) relapse, standard management now frequently incorporates prophylactic intrathecal chemotherapy once remission is achieved and the coagulopathy has resolved [9].
Frequently Asked Questions (FAQs)
What is acute promyelocytic leukemia (APL)?
APL is a subtype of acute myeloid leukemia caused by a t(15;17) chromosome swap that creates the PML-RARA fusion gene. This gene blocks young white blood cells from maturing, so they build up in the bone marrow. APL is a medical emergency because of severe bleeding risk, but it is also the most curable type of adult leukemia.
Why is APL considered a medical emergency?
The leukemia cells trigger a severe clotting and bleeding disorder called DIC. Patients can bleed catastrophically, including into the brain or lungs, within hours to days. This is why treatment with ATRA starts the moment APL is suspected, before tests confirm it.
How is APL treated today?
Most patients receive a combination of ATRA and arsenic trioxide (ATO), which forces the leukemia cells to mature and clears the abnormal protein. This largely chemotherapy-free approach is the standard of care for non-high-risk APL and produces cure rates above 90%. High-risk cases may add low-dose chemotherapy.
What is the survival rate for APL?
For patients who survive the first critical month, five-year survival exceeds 90%. The catch is the early period: in real-world populations, up to 30% of patients die within 30 days, mostly from bleeding, even though clinical trials report lower rates of 5–10%.
What is differentiation syndrome?
It is a complication of ATRA or ATO treatment, affecting roughly a quarter to a third of patients. As leukemia cells mature all at once, they release inflammatory signals that cause fever, breathing difficulty, and fluid buildup. It is treated promptly with steroids such as dexamethasone.
Can APL come back after treatment?
Relapse is uncommon with modern ATRA-ATO therapy, but it can happen. Doctors monitor for the PML-RARA gene using sensitive PCR blood tests so that any return of the leukemia can be caught and treated early.
Glossary of Related Medical Terms
- Acute promyelocytic leukemia (APL): A fast-growing blood cancer in which immature white blood cells called promyelocytes pile up in the bone marrow instead of maturing.
- Promyelocyte: An early, immature stage of a white blood cell that normally goes on to become a mature infection-fighting cell.
- t(15;17) translocation: A swap of genetic material between chromosomes 15 and 17 that creates the abnormal gene driving APL.
- PML-RARA fusion gene: The faulty gene formed by the t(15;17) swap. Its protein blocks white blood cells from maturing.
- Differentiation therapy: Treatment that forces cancer cells to grow up (mature) and die naturally, rather than poisoning them like traditional chemotherapy.
- ATRA (all-trans retinoic acid): A vitamin A–based drug that unlocks the maturation block in APL cells.
- Arsenic trioxide (ATO): A drug that helps destroy the abnormal APL protein and clear the leukemia.
- Disseminated intravascular coagulation (DIC): A dangerous state where the blood clots and bleeds uncontrollably at the same time, using up clotting factors.
- Coagulopathy: Any problem with the blood's ability to clot normally.
- Differentiation syndrome: A treatment complication where maturing leukemia cells flood the body with inflammatory signals, causing fever, fluid buildup, and breathing trouble.
- Pancytopenia: A shortage of all three main blood cell types: red cells, white cells, and platelets.
- Auer rods: Needle-like clumps seen inside leukemia cells under the microscope; stacked bundles are a clue to APL.
- Induction therapy: The first, intensive phase of treatment aimed at clearing the leukemia and achieving remission.
- Complete remission: No detectable leukemia after treatment, though monitoring continues.
Disclaimer: This article is intended for informational purposes only and is specifically targeted towards medical students. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Dores, G. M., Devesa, S. S., Curtis, R. E., Linet, M. S., & Morton, L. M. (2012). Acute leukemia incidence and patient survival among children and adults in the United States, 2001-2007. Blood, 119(1), 34–43. https://doi.org/10.1182/blood-2011-04-347872
- Abedin, S., & Altman, J. K. (2016). Acute promyelocytic leukemia: Preventing early complications and late toxicities. Hematology. American Society of Hematology Education Program, 2016(1), 10–15. https://doi.org/10.1182/asheducation-2016.1.10
- Hermsen, J., & Hambley, B. (2023). The Coagulopathy of Acute Promyelocytic Leukemia: An Updated Review of Pathophysiology, Risk Stratification, and Clinical Management. Cancers, 15(13), 3477. https://doi.org/10.3390/cancers15133477
- Sanz, M. A., Fenaux, P., Tallman, M. S., Estey, E. H., Löwenberg, B., Naoe, T., Lengfelder, E., Döhner, H., Burnett, A. K., Chen, S. J., Mathews, V., Iland, H., Rego, E., Kantarjian, H., Adès, L., Avvisati, G., Montesinos, P., Platzbecker, U., Ravandi, F., Russell, N. H., … Lo-Coco, F. (2019). Management of acute promyelocytic leukemia: updated recommendations from an expert panel of the European LeukemiaNet. Blood, 133(15), 1630–1643. https://doi.org/10.1182/blood-2019-01-894980
- Lo-Coco, F., Avvisati, G., Vignetti, M., Thiede, C., Orlando, S. M., Iacobelli, S., Ferrara, F., Fazi, P., Cicconi, L., Di Bona, E., Specchia, G., Sica, S., Divona, M., Levis, A., Fiedler, W., Cerqui, E., Breccia, M., Fioritoni, G., Salih, H. R., Cazzola, M., … Study Alliance Leukemia (2013). Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. The New England journal of medicine, 369(2), 111–121. https://doi.org/10.1056/NEJMoa1300874
- Cicconi, L., Platzbecker, U., Avvisati, G., Paoloni, F., Thiede, C., Vignetti, M., Fazi, P., Ferrara, F., Divona, M., Albano, F., Efficace, F., Sborgia, M., Di Bona, E., Breccia, M., Borlenghi, E., Cairoli, R., Rambaldi, A., Melillo, L., La Nasa, G., Fiedler, W., … Lo-Coco, F. (2020). Long-term results of all-trans retinoic acid and arsenic trioxide in non-high-risk acute promyelocytic leukemia: update of the APL0406 Italian-German randomized trial. Leukemia, 34(3), 914–918. https://doi.org/10.1038/s41375-019-0589-3
- Platzbecker, U., Adès, L., Montesinos, P., Ammatuna, E., Fenaux, P., Baldus, C., Bernardi, M., Berthon, C., Bocchia, M., Bonmati, C., Borlenghi, E., Bornhäuser, M., Carp, D., Chantepie, S., Crea, E., Divona, M., Döhner, H., Ehninger, G., Esteve Reyner, J., Frayfer, J., … SAL, AMCL-CG, AML-SG, OSHO, PETHEMA, HOVON and GIMEMA study groups (2025). Arsenic Trioxide and All-Trans Retinoic Acid Combination Therapy for the Treatment of High-Risk Acute Promyelocytic Leukemia: Results From the APOLLO Trial. Journal of clinical oncology : official journal of the American Society of Clinical Oncology, 43(29), 3160–3169. https://doi.org/10.1200/JCO-25-00535
- Park, J. H., Qiao, B., Panageas, K. S., Schymura, M. J., Jurcic, J. G., Rosenblat, T. L., Altman, J. K., Douer, D., Rowe, J. M., & Tallman, M. S. (2011). Early death rate in acute promyelocytic leukemia remains high despite all-trans retinoic acid. Blood, 118(5), 1248–1254. https://doi.org/10.1182/blood-2011-04-346437
- Abaza, Y., Kantarjian, H., Garcia-Manero, G., Estey, E., Borthakur, G., Jabbour, E., Faderl, S., O'Brien, S., Wierda, W., Pierce, S., Brandt, M., McCue, D., Luthra, R., Patel, K., Kornblau, S., Kadia, T., Daver, N., DiNardo, C., Jain, N., Verstovsek, S., … Ravandi, F. (2017). Long-term outcome of acute promyelocytic leukemia treated with all-trans-retinoic acid, arsenic trioxide, and gemtuzumab. Blood, 129(10), 1275–1283. https://doi.org/10.1182/blood-2016-09-736686
- Kayser, S., Döhner, K., Krauter, J., Köhne, C. H., Horst, H. A., Held, G., von Lilienfeld-Toal, M., Wilhelm, S., Kündgen, A., Götze, K., Rummel, M., Nachbaur, D., Schlegelberger, B., Göhring, G., Späth, D., Morlok, C., Zucknick, M., Ganser, A., Döhner, H., Schlenk, R. F., … German-Austrian AMLSG (2011). The impact of therapy-related acute myeloid leukemia (AML) on outcome in 2853 adult patients with newly diagnosed AML. Blood, 117(7), 2137–2145. https://doi.org/10.1182/blood-2010-08-301713
.jpg){kind=link}