Key Takeaways
Mast Cell Activation Syndrome or MCAS is a condition where a normal number of mast cells release their chemicals too easily, producing repeated symptoms across two or more organ systems.
- Pathophysiology and Classification ▾: MCAS involves a "lowered threshold" for activation via multiple receptor pathways (not just IgE). MCAS is classified as primary (clonal, driven by KIT mutations), secondary (reactive to another condition), or idiopathic (no clear cause).).
- Clinical Manifestations ▾: Symptoms are distinctly episodic and multisystemic (involving two or more systems simultaneously). Common presentations include flushing and hives (dermatologic), cramping and diarrhea (GI), tachycardia and hypotension (cardiovascular), wheezing (respiratory), and cognitive impairment or "brain fog" (neurologic).
- Laboratory Investigations & Differential Diagnosis ▾: Diagnosis uses three criteria: typical episodic symptoms, a documented rise in serum tryptase during a flare (baseline × 1.2 + 2 ng/mL), and response to mast-cell-directed treatment [1, 2].
- Treatment and Management ▾: Management is stepwise, beginning with trigger avoidance, then add H1 and H2 antihistamines, then mast cell stabilizers, then mediator-specific drugs, with biologics or tyrosine kinase inhibitors for severe or clonal disease.
*Click ▾ for more information
Introduction
Mast Cell Activation Syndrome (MCAS) is one of the most discussed and most contested topics in modern clinical immunology. Patients can present with flushing, hives, stomach cramps, palpitations, and brain fog that flare unpredictably. Symptoms span multiple organ systems and rarely fit a single textbook diagnosis. Many patients spend years searching for answers.
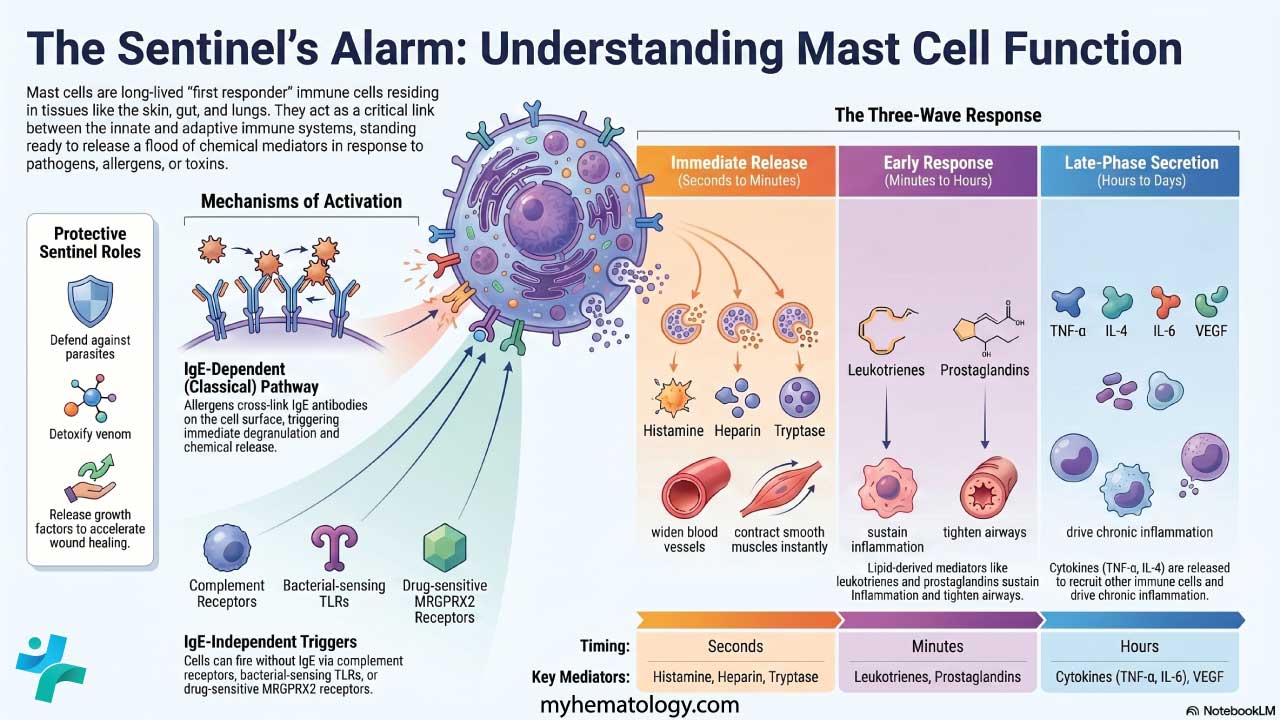
MCAS sits in the family of mast cell activation disorders. The defining problem is not how many mast cells you have — it is how they behave. The cells release their chemical mediators too easily, in response to triggers that should not provoke a reaction.
Awareness has risen sharply over the past decade. So has overdiagnosis. Studies from referral centers show that fewer than 5% of patients sent for evaluation of suspected MCAS actually meet strict diagnostic criteria [8]. A high index of suspicion is essential, but so is rigor.
The Biology of Mast Cells
Mast cells are tissue-resident immune cells. Unlike most white blood cells, they do not circulate in mature form. They settle in tissues that meet the outside world — skin, gut, airways — and act as sentinels.
Origin and maturation
Mast cells begin life in the bone marrow as CD34+/CD117+ (c-KIT) progenitors. They travel through the bloodstream as immature, agranular cells, then mature in tissue under the influence of stem cell factor (SCF). Where they settle shapes what they become. A mast cell in the gut is not identical to one in the lung.
What mast cells normally do
Mast cells are first responders. They are positioned near blood vessels, nerves, and mucosal surfaces. Their normal jobs include:
- Innate immunity: recognizing pathogens through Toll-like receptors (TLRs) and triggering inflammation.
- Adaptive immunity: cooperating with B and T cells, including in IgE-mediated allergic responses.
- Homeostasis: regulating blood vessel dilation, new vessel growth, and tissue repair.
- Neuro-immune signaling: communicating with nearby nerve endings, which helps explain the brain fog and pain often reported in MCAS.
The mediator arsenal
When activated, mast cells release a mixture of chemicals. These fall into three groups based on timing:
| Category | Key mediators | Clinical impact |
| Preformed (stored in granules) | Histamine, heparin, tryptase, chymase | Immediate vasodilation, itching, tissue swelling |
| Newly synthesized lipids | Prostaglandin D2 (PGD2), Leukotrienes (e.g., LTC4) | Airway narrowing, mucus, prolonged inflammation |
| Cytokines and chemokines | TNF-α, IL-6, IL-4 | Chronic systemic symptoms, fatigue, immune cell recruitment |
How mast cells get activated
In a healthy person, mast cells degranulate in response to a clear threat. In MCAS, the threshold is set too low. Activation pathways include:
- IgE-mediated: the classic allergic route, where IgE bound to FcεRI receptors is cross-linked by an allergen.
- Non-IgE-mediated: physical triggers (heat, cold, vibration), chemical triggers (certain drugs, fragrances, preservatives), and neuropeptides like substance P released during emotional stress.
Pathophysiology and Classification
The pathophysiology of MCAS centers on a lowered threshold for degranulation [3]. Mast cells become hyper-reactive to minor or non-specific stimuli.
Beyond IgE: the wider receptor landscape
MCAS pathophysiology involves a much broader array of receptors. This explains why triggers can be so diverse (foods, heat, stress, medications).
- FcεRI (the high-affinity IgE receptor): the classic allergic pathway.
- MRGPRX2: a more recently characterized receptor that lets mast cells respond to "pseudo-allergens," including fluoroquinolones, neuromuscular blockers, and neuropeptides.
- Toll-like receptors (TLRs): detect pathogen-associated molecular patterns, allowing IgE-independent responses to chronic infection.
- Complement receptors: respond to anaphylatoxins C3a and C5a, linking MCAS to inflammatory and autoimmune states.
Intracellular signaling
Once a receptor is engaged, a signaling cascade begins. The early phase activates Src-family tyrosine kinases such as Syk and Lyn, which trigger rapid release of preformed mediators like histamine and tryptase. The late phase activates MAP kinase and PI3K pathways, generating arachidonic-acid-derived mediators and switching on cytokine genes.
In some MCAS patients, the cell appears to sit in a pre-activated state, requiring only a small stimulus to release a large mediator burst. The precise molecular basis in non-clonal MCAS is still being worked out [hypothesis, not consensus].
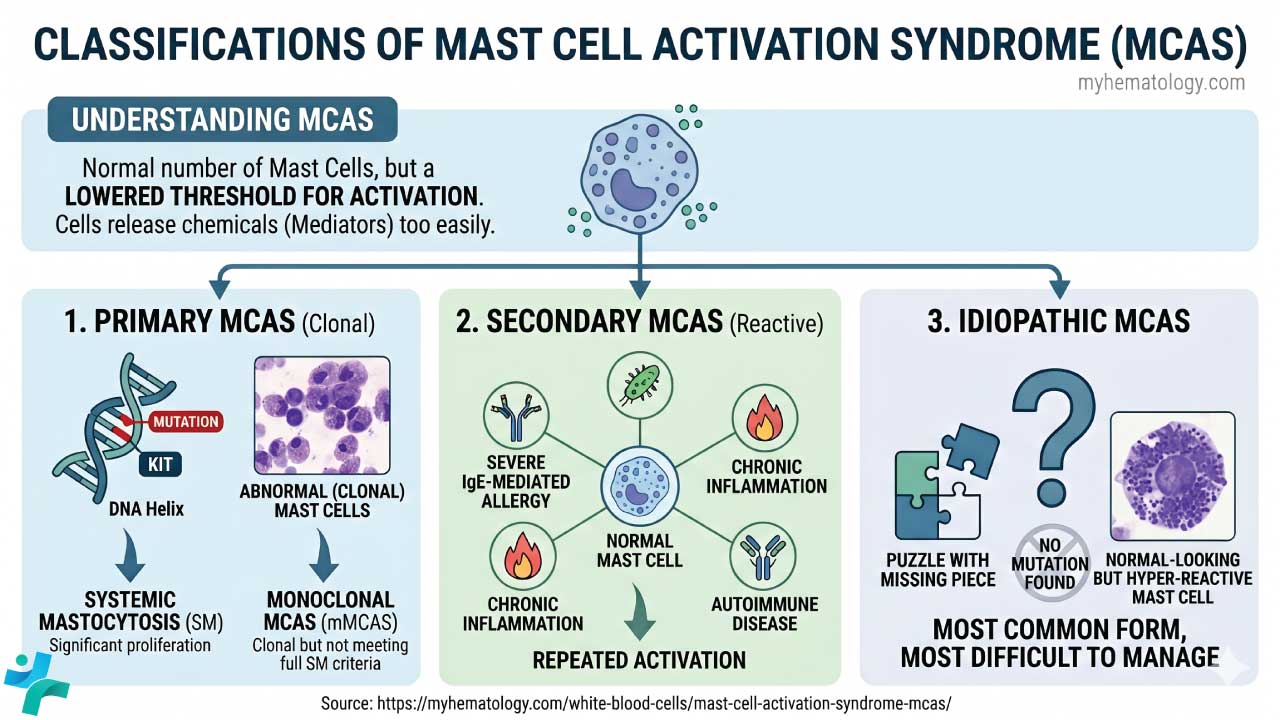
The three-tier classification
Primary MCAS (clonal). A hematological disorder where the mast cells themselves are genetically abnormal. The most common driver is a somatic KIT D816V mutation, which keeps the SCF receptor permanently switched on. When proliferation is significant, the diagnosis becomes systemic mastocytosis. When the criteria for SM are not fully met but a clonal population is detectable, it is termed monoclonal MCAS (mMCAS) [2].
Secondary MCAS (reactive). The mast cells are genetically normal but are repeatedly pushed into activation by another condition — severe IgE-mediated allergy, chronic inflammation, or autoimmune disease.
Idiopathic MCAS. The mast cells are hyper-reactive, but no clonal mutation and no clear secondary driver can be found. It is the most common form, and the most difficult to manage.
Why symptoms spread so widely
Mast cells live close to blood vessels and nerves. When they degranulate, histamine and prostaglandins cause local vasodilation and leakier capillaries — hence flushing, swelling, and sudden drops in blood pressure. Because mast cell mediators also lower nerve activation thresholds, patients can experience exaggerated pain, autonomic instability, and the cognitive sluggishness often described as brain fog.
Clinical Manifestations
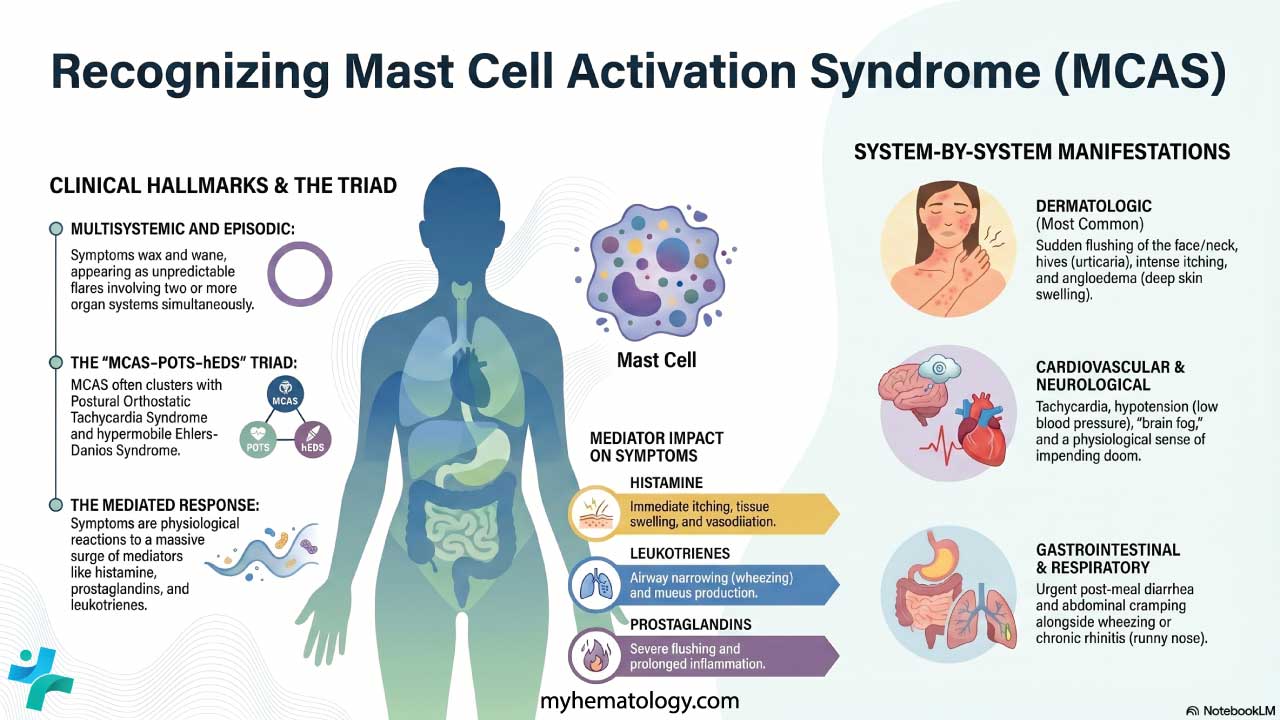
The hallmark of MCAS is symptoms that are multisystemic and episodic. Symptoms wax and wane, often appearing as flares triggered by specific exposures. The consensus criteria require two or more organ systems to be involved during a flare [1, 2].
Dermatologic (the most common)
Skin involvement appears in the large majority of MCAS patients and is often the trigger for referral.
- Flushing: sudden redness of the face, neck, and upper chest, driven mainly by histamine and PGD2.
- Pruritus and urticaria: intense itching and hives.
- Angioedema: rapid swelling of deeper skin layers, often around the eyes or lips.
- Dermatographism: a scratch produces a raised, red wheal within minutes.
Gastrointestinal
The gut has a high density of mast cells. When they degranulate, the result can be mistaken for irritable bowel syndrome.
- Cramping abdominal pain
- Urgent, post-meal diarrhea
- Nausea and vomiting
- Malabsorption and minor nutrient deficiencies with chronic activation
Cardiovascular and autonomic
These symptoms are often the most disabling and drive emergency room visits.
- Tachycardia and palpitations, especially during flares.
- Hypotension and syncope caused by systemic vasodilation during severe degranulation.
- POTS overlap: many MCAS patients also meet criteria for Postural Orthostatic Tachycardia Syndrome. The two share clinical territory but have different primary mechanisms. POTS produces tachycardia on standing without the drop in blood pressure or skin findings seen in an MCAS flare.
The MCAS–POTS–hEDS triad
A growing body of clinical experience describes a triad of MCAS, POTS, and hypermobile Ehlers-Danlos syndrome (hEDS), often clustering in the same patients. While a single unifying genetic cause remains elusive, clinical manifestations of conditions like hypertryptasemia include dysautonomia, joint hypermobility, and aberrant mast cell activation, suggesting a complex neurohormonal and connective tissue cross-talk mechanism [14]. When one of these is diagnosed, the other two are worth screening for.
Respiratory and nasopharyngeal
- Chronic rhinitis: congestion, sneezing, and runny nose without an obvious viral cause.
- Wheezing and shortness of breath, since leukotrienes are potent bronchoconstrictors.
- A sensation of throat fullness or difficulty swallowing during flares.
Neurological and psychiatric
- Brain fog: difficulty concentrating, memory lapses, and slowed thinking.
- Headaches: often migraine-like or tension-pattern.
- Acute anxiety or a sense of impending doom during a flare. This is a physiological response to a massive mediator surge and not a primary psychiatric disorder.
Pediatric MCAS
MCAS in children is uncommon and remains poorly characterized. Pediatric presentations tend to be milder than adult forms, but careful evaluation by a specialist is needed to distinguish MCAS from food allergy, chronic urticaria, and pediatric mastocytosis.
Non-IgE-mediated anaphylaxis
It is vital to distinguish classic IgE-mediated anaphylaxis from a severe MCAS flare. The clinical picture — hypotension, wheezing, hives — can look identical. However, MCAS flares may be triggered by non-specific stimuli (heat, vibration) and proceed through receptors other than IgE [7]. The older term "anaphylactoid" has been retired in favor of "non-IgE-mediated anaphylaxis" or "non-immunologic anaphylaxis."
Laboratory Investigations
Diagnosing MCAS is difficult because mast cell mediators have very short half-lives in circulation. A negative test does not rule out the condition when clinical suspicion is high and sample handling is suboptimal [1].
The unreliability of plasma histamine
While frequently ordered in primary care, routine plasma histamine blood tests are virtually useless for diagnosing MCAS in standard clinical settings. Histamine degrades extremely rapidly in the bloodstream—with a half-life of roughly 1 to 2 minutes—yielding high rates of false-negative results. Consequently, current diagnostic frameworks strongly prioritize serum tryptase and 24-hour urinary metabolites over plasma histamine testing [7].
Serum tryptase: the most specific marker
Tryptase is the most specific blood marker of mast cell degranulation, but a single random level is rarely useful.
- Baseline tryptase: measured when the patient is asymptomatic.
- Event tryptase: drawn within 30 to 120 minutes of symptom onset during a flare.
- The consensus formula: a diagnosis is supported when event tryptase rises by at least 20% over the patient's baseline, plus 2 ng/mL [2, 3].
Example: If baseline tryptase is 5 ng/mL, the event tryptase must reach at least (5 × 1.2) + 2 = 8 ng/mL.
24-hour urine mediators
When serum tryptase is normal (as it often is in MCAS), urine mediator metabolites are the next step.
- N-methylhistamine, a metabolite of histamine
- 11β-prostaglandin F2α (11β-PGF2α), a metabolite of PGD2
- Leukotriene E4 (LTE4), a leukotriene metabolite
Critical handling point
Pre-analytical mishandling is a primary cause of false-negative results. Samples must be kept strictly refrigerated or on ice from the moment of first voiding until lab processing. Modern clinical protocols advise patients to speak directly with the laboratory manager prior to dropping off the 24-hour collection jug to ensure the cold chain is maintained immediately upon handover [15].
Hematological and histopathological studies
When primary (clonal) MCAS is suspected:
- Bone marrow aspiration and biopsy: looking for mast cell aggregates, spindle-shaped morphology, and aberrant expression of CD25 or CD2.
- Genetic testing: high-sensitivity assays for the KIT D816V mutation in peripheral blood or bone marrow.
- GI biopsies: CD117 (c-KIT) staining during endoscopy can quantify mucosal mast cells. Reference ranges vary by institution.
Testing for hereditary alpha-tryptasemia
When baseline tryptase is persistently elevated without an obvious cause, TPSAB1 copy number testing is now recommended [5]. HαT is an autosomal dominant genetic trait affecting roughly 5–7% of the Western population. Rather than just a differential diagnosis, HαT is now recognized as a major genetic disease modifier of mast cell disorders. The presence of extra α-tryptase alleles promotes the formation of unique heterotetramers that lower the activation threshold of mast cells, thereby amplifying MCAS symptoms and significantly raising the risk of severe anaphylaxis [5,9].
Differential Diagnosis
MCAS is a multisystem mimic. Common and dangerous conditions must be excluded first.
Systemic mastocytosis (SM)
The defining contrast: SM is a proliferative disorder with an excess of mast cells, while MCAS is an activation disorder with a normal mast cell count. Baseline tryptase in SM is usually chronically elevated (often above 20 ng/mL); in MCAS it is typically normal between flares [2].
Hereditary alpha-tryptasemia (HαT)
HαT is a common genetic trait with extra copies of TPSAB1. Baseline tryptase is chronically raised, and patients may have symptoms that overlap with MCAS. HαT is now recognized as both a differential and a modifier — a person can have HαT, MCAS, or both [5].
Carcinoid syndrome
Overlap: flushing and diarrhea. The difference: carcinoid is driven by serotonin-secreting neuroendocrine tumors. Look for elevated 24-hour urinary 5-HIAA, and note that hives and angioedema are typical of MCAS but not carcinoid.
Pheochromocytoma
Overlap: episodic tachycardia, hypertension, and anxiety-like spells. The difference: catecholamine-secreting adrenal tumors. Plasma or urinary metanephrines are the key test.
Other differentials
- POTS (Postural Orthostatic Tachycardia Syndrome): frequently co-exists with MCAS but does not produce hives or GI distress on its own.
- Classic IgE-mediated anaphylaxis: usually has one identifiable trigger and is not a chronic multisystemic syndrome.
- Vasovagal syncope: lacks mediator-driven features like flushing and urticaria.
| Condition | Overlapping Symptoms with MCAS | Key Differentiating Features | Primary Diagnostic Test(s) |
| Systemic Mastocytosis (SM) | Flushing, hives, anaphylaxis, GI upset, tachycardia, brain fog. | SM is a proliferative cancer-like disorder (too many mast cells), whereas MCAS is an activation disorder (normal number, but hyper-reactive). | Bone marrow biopsy (looking for mast cell aggregates), baseline serum tryptase (usually >20 ng/mL), KIT D816V mutation testing. |
| HαT | Flushing, GI issues, dysautonomia, increased risk of anaphylaxis. | HαT is a genetic trait (extra gene copies), not an activation disorder itself, though it acts as a disease modifier and often co-occurs with MCAS. | TPSAB1 gene copy number testing; baseline tryptase is consistently >8 ng/mL. |
| Carcinoid Syndrome | Severe facial/chest flushing, sudden diarrhea, tachycardia, wheezing. | Driven by serotonin from neuroendocrine tumors. Unlikely to cause hives (urticaria) or lip/eye swelling (angioedema), which are common in MCAS. | 24-hour urine test for 5-HIAA (a serotonin metabolite); serum Chromogranin A. |
| Pheochromocytoma | Racing heart (tachycardia), anxiety/panic-like spells, sweating, headaches. | Driven by adrenaline/noradrenaline from an adrenal tumor. Typically causes severe hypertension (high blood pressure) during spells, whereas MCAS flares often cause hypotension (low blood pressure). | Plasma free metanephrines or 24-hour urine fractionated metanephrines. |
| POTS | Tachycardia, lightheadedness, brain fog, fainting, GI upset. | Symptoms are strictly triggered by gravity/posture (standing up) and improve when lying down. Does not cause hives or allergic skin flushing. (Note: Frequently co-occurs with MCAS). | Tilt table test or 10-minute active stand test. |
| Classic IgE-Mediated Allergy / Anaphylaxis | Hives, throat swelling, wheezing, sudden drop in blood pressure. | Has a clear, consistent, identifiable trigger (e.g., peanuts, bee sting). It is not an unpredictable, chronic, daily multisystemic syndrome. | Allergen-specific IgE blood tests (RAST) or skin prick testing. |
| Vasovagal Syncope | Fainting, dizziness, nausea, sweating. | Triggered by stress, pain, or prolonged standing. Lacks mediator-driven features like hives, intense itching, or deep red flushing. | Clinical history; occasionally a tilt table test. |
| Chronic Spontaneous Urticaria (CSU) | Chronic hives, severe itching, occasional angioedema. | Symptoms are primarily limited to the skin. Lacks the severe multi-organ involvement (GI, cardiovascular, neurological) seen during an MCAS flare. | Clinical diagnosis; exclusion of other systemic causes. |
Treatment and Management
MCAS is rarely cured. The goal is to stabilize the mast cells, block the effects of the mediators they release, and improve quality of life. A stepwise approach is the most practical framework.
Step 1: Trigger avoidance and lifestyle modification
Identifying personal triggers comes first.
- Common physical triggers include heat, cold, sudden temperature changes, friction, and emotional stress. Alcohol is a frequent culprit.
- A low-histamine diet helps some patients. Foods to consider limiting include fermented products, aged cheeses, and certain processed meats.
- The "filler" ingredients or excipients in tablets (such as dyes, lactose, and preservatives) can frequently trigger reactions even when the active drug is well-tolerated. Therefore, when initiating pharmacotherapy, a strict stepwise approach is critical: new medications or dose increases should be introduced one at a time, allowing for an adequate trial period to monitor for adverse effects and objective clinical improvement [13]. Switching to a compounded, filler-free formulation may resolve excipient-driven flares.
Step 2: Baseline receptor blockade
H1 and H2 antihistamines form the first medical line of defense [7].
- H1 antihistamines: non-sedating second-generation agents (such as cetirizine, loratadine, and fexofenadine) are the preferred first-line intervention [13]. Under medical supervision, MCAS patients often require a targeted up-dosing protocol where the standard daily dose is increased up to four times, an approach extrapolated from international chronic urticaria guidelines.
- H2 antihistamines: famotidine is the usual choice. H2 receptors are present on mast cells and blood vessels, so combining H1 and H2 blockade produces a more complete histamine effect than either alone.
Step 3: Mast cell stabilizers
When receptor blockade is not enough, the next step is to prevent degranulation.
- Oral cromolyn sodium: poorly absorbed systemically, which is precisely why it works well in the gut. A good choice for patients whose dominant symptoms are abdominal pain and diarrhea.
- Ketotifen: combines potent H1 antihistamine activity with systemic mast cell stabilization.
Step 4: Targeting other mediators
When flares persist, secondary mediators are targeted.
- Leukotriene receptor antagonists (montelukast) help with respiratory symptoms and some skin manifestations.
- Low-dose aspirin can suppress PGD2 production in patients whose flushing is prostaglandin-driven. Because aspirin is itself a potential trigger, this is initiated only under specialist supervision, usually after a controlled drug challenge.
Step 5: Advanced and biological therapies
For refractory or severe disease:
- Omalizumab (anti-IgE): a monoclonal antibody that binds free IgE, lowering FcεRI activation and raising the threshold for flares.
- Anti-Siglec-8 antibodies (e.g., lirentelimab): An emerging frontier in mast cell-targeting therapies. Lirentelimab is a monoclonal antibody against sialic acid-binding immunoglobulin-like lectin (Siglec)-8 that selectively inhibits mast cell activation and depletes eosinophils. It has shown promising results in clinical trials for both antihistamine-resistant chronic urticaria [10] and indolent systemic mastocytosis, demonstrating improvements in symptom severity and quality of life [11,12]. It offers therapeutic potential for severe disease where standard treatments fail.
- Tyrosine kinase inhibitors (TKIs): in clonal mast cell disease driven by KIT D816V, TKIs target the hyperactive receptor directly. Avapritinib (Ayvakit) was approved by the FDA in June 2021 for advanced systemic mastocytosis and in May 2023 for indolent systemic mastocytosis based on the phase 2 PIONEER trial [4]. PIONEER showed significant improvements in symptom scores, mast cell burden, and quality of life compared with placebo. This represents the first disease-modifying therapy for a previously supportive-care-only condition. Midostaurin remains an option in advanced systemic mastocytosis.
Emergency management
Every patient with a history of severe reactions should carry epinephrine auto-injectors and have a written anaphylaxis action plan. A medical alert ID is strongly recommended.
Antihistamines and steroids are too slow for an acute, life-threatening flare. Epinephrine is the only agent that rapidly reverses systemic vasodilation and bronchoconstriction. Patients and caregivers should rehearse the action plan, because in the moment, hesitation costs time.
Pharmacotherapy at a glance
| Target | Medication | Primary indication |
| Histamine H1 | Cetirizine, fexofenadine | Itching, hives, flushing |
| Histamine H2 | Famotidine | GI upset, systemic stabilizing |
| Degranulation | Cromolyn, ketotifen | GI symptoms, overall stabilization |
| Leukotrienes | Montelukast | Wheezing, congestion, skin |
| Prostaglandins | Aspirin | Severe flushing |
| IgE pathway | Omalizumab | Refractory multisystemic flares |
| KIT D816V | Avapritinib | Clonal MCAS and systemic mastocytosis |
Prognosis and Quality of Life
MCAS is rarely fatal. The burden of disease, however, can be heavy, and long-term clinical partnership is essential.
Long-term outlook
The prognosis depends on the variant.
- Secondary and idiopathic MCAS: do not affect life expectancy. The goal is symptom control. Many patients lead near-normal lives once triggers are identified and medications are optimized.
- Primary (clonal) MCAS: prognosis depends on whether the condition remains indolent or progresses toward systemic mastocytosis. Most clonal MCAS stays stable for decades. Annual tryptase levels and complete blood counts are routine for monitoring.
The patient experience
MCAS scores poorly on quality-of-life measures, largely because of the unpredictability of flares.
- The invisible illness: patients look healthy between flares but live in a state of constant vigilance. Past emergency room experiences, where symptoms were dismissed, often leave lasting medical trauma.
- Restricted lives: severe MCAS can force narrow safe-food lists and avoidance of fragranced or chemical-rich environments. This affects work, social life, and identity.
- The psychological toll: chronic fatigue, brain fog, and the physiological anxiety produced by mediator surges can be misread as primary psychiatric illness. Validating the physical reality of these symptoms is part of effective care.
Multidisciplinary management
Because MCAS crosses organ boundaries, care works best as a team effort:
- Immunology or hematology: oversees mast cell management and clonal workup.
- Gastroenterology: addresses chronic GI symptoms and malabsorption.
- Nutrition: balances dietary restrictions with adequate intake.
- Psychology and counseling: helps patients cope with unpredictability and reframe mediator-driven anxiety.
Success in MCAS is not measured by symptom elimination but by reducing the frequency and severity of flares. Moving from three flares a week to one flare a month is a meaningful clinical victory.
Resources for Patients and Caregivers
- The Mast Cell Disease Society (TMS): patient-led organization with educational materials and clinician directories (tmsforacure.org).
- NIAID Laboratory of Allergic Diseases: research and patient information at niaid.nih.gov.
- American Academy of Allergy, Asthma & Immunology (AAAAI): clinician-facing guidelines and patient resources at aaaai.org.
Frequently Asked Questions
What is MCAS in simple terms?
MCAS, or Mast Cell Activation Syndrome, is a condition where mast cells release histamine and other chemicals when they should not. This causes flushing, hives, stomach upset, low blood pressure, and brain fog that come and go. The number of mast cells is usually normal; the problem is how easily they get activated.
Is MCAS the same as systemic mastocytosis?
No. Systemic mastocytosis is a blood disorder with too many abnormal mast cells. MCAS involves a normal count of mast cells that are over-reactive. The two overlap when a KIT D816V mutation is present, but diagnosis and treatment differ.
Can a normal tryptase level rule out MCAS?
No. Tryptase has a short half-life and must be measured within 30 to 120 minutes of a flare. Some flares are driven mainly by prostaglandins or leukotrienes, which do not raise tryptase. A normal result does not exclude MCAS when the clinical picture fits.
Is MCAS curable?
There is no cure as of 2026. Most patients gain meaningful control by avoiding triggers and combining H1 and H2 antihistamines, mast cell stabilizers, and mediator-specific drugs. For patients with the KIT D816V mutation and indolent systemic mastocytosis, the FDA approved avapritinib (Ayvakit) in May 2023, offering the first disease-modifying option.
What is the link between MCAS, POTS, and Ehlers-Danlos syndrome?
The three often cluster together, especially in young women. The biological basis is still being worked out, but mast cell mediators may affect blood vessel tone and connective tissue. When one is diagnosed, screening for the other two is reasonable.
Should I get tested for hereditary alpha-tryptasemia?
If your baseline tryptase is persistently above 8 ng/mL without another explanation, TPSAB1 copy number testing is worth discussing with a specialist.
Glossary of Related Medical Terms
- Anaphylaxis — A severe, rapid-onset allergic reaction affecting multiple organ systems. Can be life-threatening without epinephrine.
- Angioedema — Swelling of the deeper layers of the skin, often around the eyes, lips, or throat.
- Avapritinib — A targeted drug that blocks the abnormal KIT D816V protein driving clonal mast cell disease.
- Cytokine — A small signaling protein released by immune cells to coordinate inflammation.
- Degranulation — The process where a mast cell empties its storage granules, releasing chemicals like histamine into surrounding tissue.
- Dermatographism — A skin reaction where light scratching causes raised, red lines or welts within minutes.
- Hereditary alpha-tryptasemia (HαT) — An inherited trait where a person has extra copies of the TPSAB1 gene, leading to higher baseline tryptase levels.
- Histamine — A chemical released by mast cells that causes itching, redness, swelling, and dropped blood pressure.
- KIT D816V — A specific mutation in the KIT gene that switches the receptor permanently "on," causing mast cells to activate or multiply abnormally.
- Leukotrienes — Fat-derived signaling molecules that cause airway tightening and mucus production.
- Mast cell — A tissue-based immune cell that stores potent chemicals and releases them in response to threats or, in MCAS, to inappropriate triggers.
- MCAS (Mast Cell Activation Syndrome) — A disorder where a normal number of mast cells are abnormally easy to activate, releasing chemicals that cause repeated multi-system symptoms.
- Prostaglandin D2 (PGD2) — A lipid mediator released by mast cells that drives flushing and bronchoconstriction.
- Systemic mastocytosis (SM) — A disease where the body produces too many abnormal mast cells, usually due to a KIT mutation.
- Tryptase — An enzyme stored in mast cells; serum levels rise during activation and are used as a key diagnostic marker.
Disclaimer: This article is intended for informational purposes only and is specifically targeted towards medical students. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Valent, P., Akin, C., Bonadonna, P., Hartmann, K., Brockow, K., Niedoszytko, M., Nedoszytko, B., Siebenhaar, F., Sperr, W. R., Oude Elberink, J. N. G., Butterfield, J. H., Alvarez-Twose, I., Sotlar, K., Reiter, A., Kluin-Nelemans, H. C., Hermine, O., Gotlib, J., Broesby-Olsen, S., Orfao, A., Horny, H. P., … Metcalfe, D. D. (2019). Proposed Diagnostic Algorithm for Patients with Suspected Mast Cell Activation Syndrome. The journal of allergy and clinical immunology. In practice, 7(4), 1125–1133.e1. https://doi.org/10.1016/j.jaip.2019.01.006
- Valent, P., Akin, C., Hartmann, K., Alvarez-Twose, I., Brockow, K., Hermine, O., Niedoszytko, M., Schwaab, J., Lyons, J. J., Carter, M. C., Elberink, H. O., Butterfield, J. H., George, T. I., Greiner, G., Ustun, C., Bonadonna, P., Sotlar, K., Nilsson, G., Jawhar, M., Siebenhaar, F., … Metcalfe, D. D. (2021). Updated Diagnostic Criteria and Classification of Mast Cell Disorders: A Consensus Proposal. HemaSphere, 5(11), e646. https://doi.org/10.1097/HS9.0000000000000646
- Valent, P., Hartmann, K., Bonadonna, P., Niedoszytko, M., Triggiani, M., Arock, M., & Brockow, K. (2022). Mast Cell Activation Syndromes: Collegium Internationale Allergologicum Update 2022. International archives of allergy and immunology, 183(7), 693–705. https://doi.org/10.1159/000524532
- Gotlib, J., Castells, M., Elberink, H. O., Siebenhaar, F., Hartmann, K., Broesby-Olsen, S., George, T. I., Panse, J., Alvarez-Twose, I., Radia, D. H., Tashi, T., Bulai Livideanu, C., Sabato, V., Heaney, M., Van Daele, P., Cerquozzi, S., Dybedal, I., Reiter, A., Pongdee, T., Barete, S., … Maurer, M. (2023). Avapritinib versus Placebo in Indolent Systemic Mastocytosis. NEJM evidence, 2(6), EVIDoa2200339. https://doi.org/10.1056/EVIDoa2200339
- Lyons, J. J., Yu, X., Hughes, J. D., Le, Q. T., Jamil, A., Bai, Y., Ho, N., Zhao, M., Liu, Y., O'Connell, M. P., Trivedi, N. N., Nelson, C., DiMaggio, T., Jones, N., Matthews, H., Lewis, K. L., Oler, A. J., Carlson, R. J., Arkwright, P. D., Hong, C., … Milner, J. D. (2016). Elevated basal serum tryptase identifies a multisystem disorder associated with increased TPSAB1 copy number. Nature genetics, 48(12), 1564–1569. https://doi.org/10.1038/ng.3696
- Akin, C., Valent, P., & Metcalfe, D. D. (2010). Mast cell activation syndrome: Proposed diagnostic criteria. The Journal of allergy and clinical immunology, 126(6), 1099–104.e4. https://doi.org/10.1016/j.jaci.2010.08.035
- Weiler, C. R., Austen, K. F., Akin, C., Barkoff, M. S., Bernstein, J. A., Bonadonna, P., Butterfield, J. H., Carter, M., Fox, C. C., Maitland, A., Pongdee, T., Mustafa, S. S., Ravi, A., Tobin, M. C., Vliagoftis, H., & Schwartz, L. B. (2019). AAAAI Mast Cell Disorders Committee Work Group Report: Mast cell activation syndrome (MCAS) diagnosis and management. The Journal of allergy and clinical immunology, 144(4), 883–896. https://doi.org/10.1016/j.jaci.2019.08.023
- Zaghmout, T., Maclachlan, L., Bedi, N., & Gülen, T. (2024). Low Prevalence of Idiopathic Mast Cell Activation Syndrome Among 703 Patients With Suspected Mast Cell Disorders. The journal of allergy and clinical immunology. In practice, 12(3), 753–761. https://doi.org/10.1016/j.jaip.2023.11.041
- Giannetti, M. P., Weller, E., Bormans, C., Novak, P., Hamilton, M. J., & Castells, M. (2021). Hereditary alpha-tryptasemia in 101 patients with mast cell activation-related symptomatology including anaphylaxis. Annals of allergy, asthma & immunology : official publication of the American College of Allergy, Asthma, & Immunology, 126(6), 655–660. https://doi.org/10.1016/j.anai.2021.01.016
- Altrichter, S., Staubach, P., Pasha, M., et al. (2022). An open-label, proof-of-concept study of lirentelimab for antihistamine-resistant chronic spontaneous and inducible urticaria. Journal of Allergy and Clinical Immunology, 149, 1683-1690.e7. https://doi.org/10.1016/j.jaci.2021.12.772
- Siebenhaar, F., Altrichter, S., Bonnekoh, H., et al. (2023). Safety and efficacy of lirentelimab in patients with refractory indolent systemic mastocytosis: a first-in-human clinical trial. British Journal of Dermatology, 189, 511-519. https://doi.org/10.1093/bjd/ljad191
- Sabato, V., Beyens, M., Toscano, A., Van Gasse, A., & Ebo, D. G. (2024). Mast Cell–Targeting Therapies in Mast Cell Activation Syndromes. Current Allergy and Asthma Reports, 24, 63-71. https://doi.org/10.1007/s11882-023-01123-9
- Nurmatov, U. B., Rhatigan, E., Simons, F. E. R., & Sheikh, A. (2015). H1‐antihistamines for primary mast cell activation syndromes: a systematic review. Allergy, 70, 1052-1061. https://doi.org/10.1111/all.12672
- Seneviratne, S. L., Maitland, A., & Afrin, L. (2017). Mast cell disorders in Ehlers–Danlos syndrome. American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 175, 226-236. https://doi.org/10.1002/ajmg.c.31555
- Afrin, L. B., Butterfield, J. H., Raithel, M., & Molderings, G. J. (2016). Often seen, rarely recognized: mast cell activation disease—a guide to diagnosis and therapeutic options. Annals of Medicine, 48(3), 190-201. https://doi.org/10.3109/07853890.2016.1161231