Key Takeaways
Thalassemia is a group of inherited blood disorders characterized by the reduced or absent production of hemoglobin, the protein in red blood cells that carries oxygen.
Pathogenesis ▾
- Caused by genetic mutations affecting globin chain production (alpha or beta).
- Imbalance of globin chains leads to ineffective erythropoiesis and hemolytic anemia.
Clinical Manifestations ▾
- Common symptoms include fatigue, weakness, pallor, and jaundice.
- Complications include iron overload, splenomegaly, bone abnormalities, and heart problems.
Diagnosis ▾
- Based on blood tests, including complete blood count (low Hb, low MCV, low MCH), peripheral blood smear (hypochromic microcytic red cells), and hemoglobin electrophoresis/HPLC (decreased HbA and increased HbF).
- Genetic testing confirms the diagnosis.
Treatment ▾
- Includes blood transfusions, iron chelation therapy, folic acid supplementation, and bone marrow transplantation.
- Gene therapy is an emerging treatment option.
Prevention ▾
- Genetic counseling, prenatal diagnosis, newborn screening, and carrier screening programs.
- Public health awareness and education.
*Click ▾ for more information
Introduction
Thalassemia is a group of inherited blood disorders characterized by the reduced or absent production of hemoglobin, the protein in red blood cells that carries oxygen. This reduction in hemoglobin leads to anemia, which is a condition where the blood doesn’t have enough healthy red blood cells.
Brief Historical Overview
The history of thalassemia is intertwined with the history of medicine and our understanding of blood disorders.
- Early Recognition: While the exact date of the first recognition of thalassemia is unclear, descriptions of similar conditions can be traced back centuries. Ancient Greek physicians, for instance, described anemic conditions.
- Cooley’s Anemia: A significant milestone came in the early 20th century when Dr. Thomas Benton Cooley, an American pediatrician, described a severe form of anemia in children of Mediterranean descent. This condition, later termed beta-thalassemia major, was initially known as Cooley’s anemia.
- Naming of Thalassemia: The term “thalassemia” was coined later, derived from the Greek words “thalassa” meaning “sea” and “emia” meaning “blood.” This reflects the geographical distribution of the disorder, predominantly in countries bordering the Mediterranean Sea.
- Genetic Understanding: The genetic basis of thalassemia was elucidated in the mid-20th century, leading to a deeper understanding of its inheritance patterns and the development of diagnostic tests.
- Modern Management: Advances in medical technology, including blood transfusions, iron chelation therapy, and bone marrow transplantation, have significantly improved the management and prognosis of thalassemia.
It’s important to note that while the understanding of thalassemia has progressed significantly, the condition remains a significant health challenge in many parts of the world.
Global Distribution of Thalassemia
Thalassemia is predominantly found in populations around the Mediterranean Sea, the Middle East, Indian subcontinent, Southeast Asia, and parts of Africa. These regions are often referred to as the “thalassemia belt.” However, due to increased global migration, the distribution of thalassemia is becoming more widespread. It’s now observed in various populations worldwide, including those with ancestral origins from the thalassemia belt.
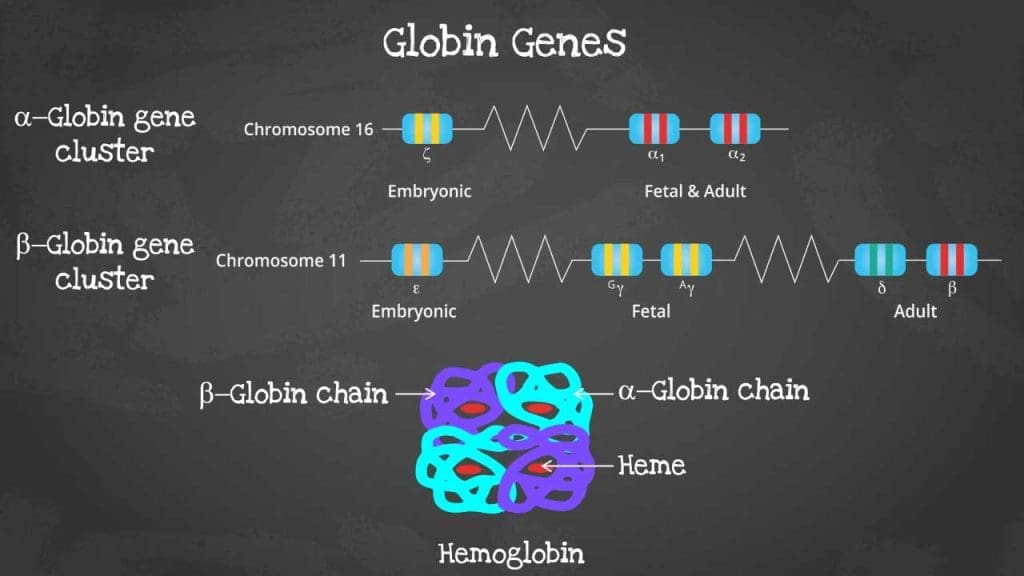
Structure of Hemoglobin
Hemoglobin is a complex protein found in red blood cells.
It is composed of four subunits, each containing a globin chain and a heme group.
- Globin chains: These are polypeptide chains that form the protein part of hemoglobin. There are different types of globin chains, including alpha, beta, delta, and gamma, which combine in various ways to form different types of hemoglobin.
- Heme group: This is a non-protein component that contains an iron atom. The iron atom is crucial for binding oxygen.
Function of Hemoglobin
The primary function of hemoglobin is to transport oxygen from the lungs to the tissues and carbon dioxide from the tissues back to the lungs.
- Oxygen transport: Hemoglobin binds oxygen in the lungs, forming oxyhemoglobin. This oxygen-rich blood is then transported to the tissues, where oxygen is released for cellular respiration.
- Carbon dioxide transport: Hemoglobin also plays a role in transporting carbon dioxide, although to a lesser extent than oxygen. Carbon dioxide binds to the amino acid residues of the globin chains, forming carbaminohemoglobin.
In addition to oxygen and carbon dioxide transport, hemoglobin also contributes to the acid-base balance of the blood.
Pathogenesis of Thalassemia
Thalassemia is a complex disorder arising from an imbalance in globin chain synthesis.
Imbalance in Globin Chain Production
- Genetic Basis: Thalassemia is caused by mutations in the genes responsible for producing globin chains, either alpha or beta.
- Reduced or Absent Globin: These mutations lead to reduced or absent production of one or both types of globin chains.
- Imbalance: The imbalance in globin chain production is the core issue. Excess of one chain (alpha or beta) occurs due to the deficit in the other.
Formation of Hemoglobin Tetramers
- Normal Hemoglobin: Normally, two alpha and two beta chains combine to form a stable hemoglobin tetramer.
- Abnormal Hemoglobin: In thalassemia, the imbalance in globin chains prevents normal tetramer formation.
- Unstable Hemoglobin: Excess globin chains form unstable intermediate complexes.
Precipitated Globin Chains
- Cellular Damage: These unstable complexes precipitate within the red blood cell, damaging the cell membrane.
- Ineffective Erythropoiesis: This damage leads to premature destruction of red blood cells in the bone marrow before they mature.
- Hemolytic Anemia: The released immature red blood cells and those damaged by the precipitated globin chains are rapidly destroyed by the spleen, leading to hemolytic anemia.
Secondary Effects
- Iron Overload: Increased red blood cell turnover and ineffective erythropoiesis lead to iron overload.
- Splenomegaly: The spleen becomes enlarged due to increased workload in filtering damaged red blood cells.
- Bone Marrow Expansion: The bone marrow tries to compensate for decreased red blood cell production, leading to bone abnormalities.
In summary, thalassemia is a cascade of events initiated by an imbalance in globin chain production. This imbalance leads to red blood cell damage, hemolysis, and subsequent complications.
Genetic basis of thalassemia
Thalassemia is an inherited blood disorder resulting from mutations in the genes responsible for producing hemoglobin. These mutations lead to reduced or absent production of globin chains, causing an imbalance in hemoglobin synthesis.
Inheritance Pattern
Thalassemia follows an autosomal recessive inheritance pattern. This means both parents must carry the defective gene for their offspring to develop the disease. Carriers, with only one defective gene, are usually asymptomatic.
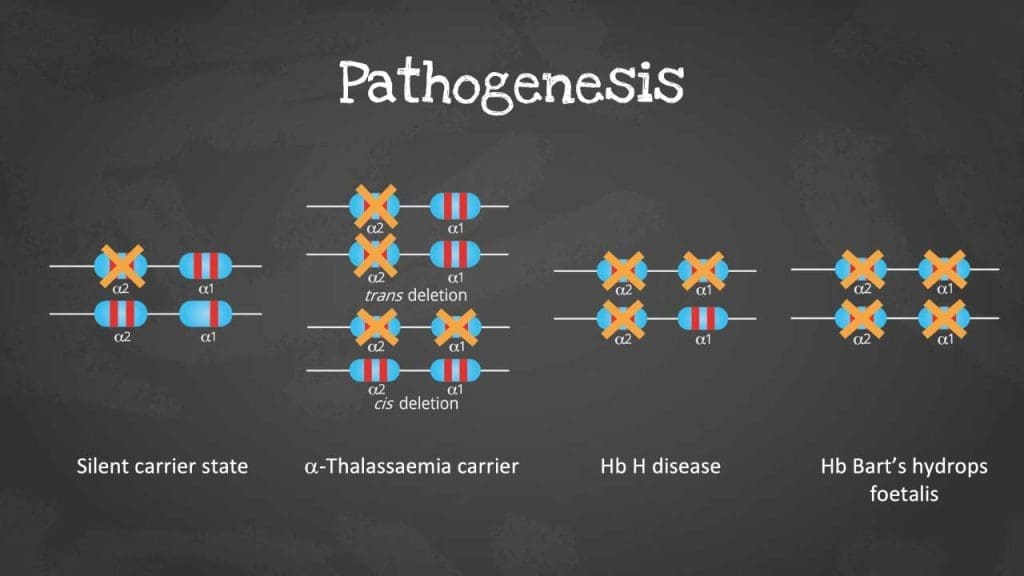
Alpha-Thalassemia
- Types
- α⁰-thalassemia: Complete absence of alpha-globin chain production.
- α⁺-thalassemia: Reduced production of alpha-globin chain.
- Gene Deletions and Mutations
- Most commonly caused by deletions of one, two, three, or all four alpha-globin genes on chromosome 16.
- Silent carrier: Deletion of one alpha-globin gene, usually asymptomatic.
- Alpha-thalassemia trait: Deletion of two alpha-globin genes, mild anemia.
- Hemoglobin H disease: Deletion of three alpha-globin genes, moderate to severe anemia.
- Hydrops fetalis: Deletion of all four alpha-globin genes, lethal condition..
- Other rare mutations affecting the alpha-globin gene structure can also lead to alpha-thalassemia.
Beta-Thalassemia
- Types
- β⁰-thalassemia: Complete absence of beta-globin chain production.
- β⁺-thalassemia: Reduced production of beta-globin chain.
- Gene Mutations
- Beta-thalassemia is caused by a wide variety of mutations in the beta-globin gene. These mutations can affect the structure or production of the beta-globin chain.
- Examples of beta-thalassemia mutations
- IVS-II-654 (C->T) mutation: A common mutation found in many populations, leading to beta-thalassemia major.
- Codons 8/9 deletion: A deletion of two nucleotides, leading to a shortened and unstable beta-globin chain.
- IVS-I-1 (G->T) mutation: A mutation in the splice acceptor site of intron 1, leading to abnormal splicing of the beta-globin mRNA.
- Numerous other point mutations can lead to beta-thalassemia, varying in severity based on their impact on beta-globin production.
Rare Types of Thalassemia
- Delta beta-thalassemia: Reduced production of both delta and beta globin chains, leading to a milder form of thalassemia compared to beta-thalassemia major.
- Gamma delta beta-thalassemia: Reduced production of gamma, delta, and beta globin chains, resulting in a more severe form of thalassemia.
Clinical Manifestations
General Clinical Features
Thalassemia is a heterogeneous group of disorders, but there are some common clinical features.
- Anemia: The hallmark of thalassemia is anemia, characterized by a reduced number of red blood cells or decreased hemoglobin levels.
- Fatigue: Due to anemia, patients often experience fatigue, weakness, and shortness of breath.
- Pallor: The skin and mucous membranes may appear pale due to decreased hemoglobin.
- Jaundice: This can occur due to increased breakdown of red blood cells.
- Splenomegaly: The spleen often becomes enlarged as it works harder to filter damaged red blood cells.
- Hepatomegaly: The liver may also be enlarged due to increased workload.
- Bone Changes: In severe cases, bone changes may occur, such as frontal bossing and skeletal deformities.
Specific Clinical Features Based on Thalassemia Type
The severity of symptoms varies widely depending on the type of thalassemia.
Alpha-Thalassemia
- Silent carrier: Usually asymptomatic.
- Alpha-thalassemia trait: Mild microcytic anemia.
- Hemoglobin H disease: Moderate to severe anemia, with jaundice, splenomegaly, and fatigue.
- Hydrops fetalis: Severe anemia leading to fetal death.
Beta-Thalassemia
- Beta-thalassemia trait: Usually asymptomatic or mild microcytic anemia.
- Beta-thalassemia intermedia: Moderate anemia, with variable symptoms.
- Beta-thalassemia major: Severe anemia requiring regular blood transfusions, with complications such as iron overload, bone marrow expansion, and organ damage.
Important note: The clinical manifestations of thalassemia can be influenced by various factors, including the specific genetic mutation, the interaction with other genetic factors, and environmental conditions.
Diagnosis
Laboratory investigations play a crucial role in diagnosing thalassemia and differentiating it from other microcytic anemias.
Basic Hematological Tests
- Complete Blood Count (CBC): Essential for assessing red blood cell indices (MCV, MCH, MCHC), which are typically decreased in thalassemia.
- Reticulocyte count: To evaluate bone marrow response to anemia.
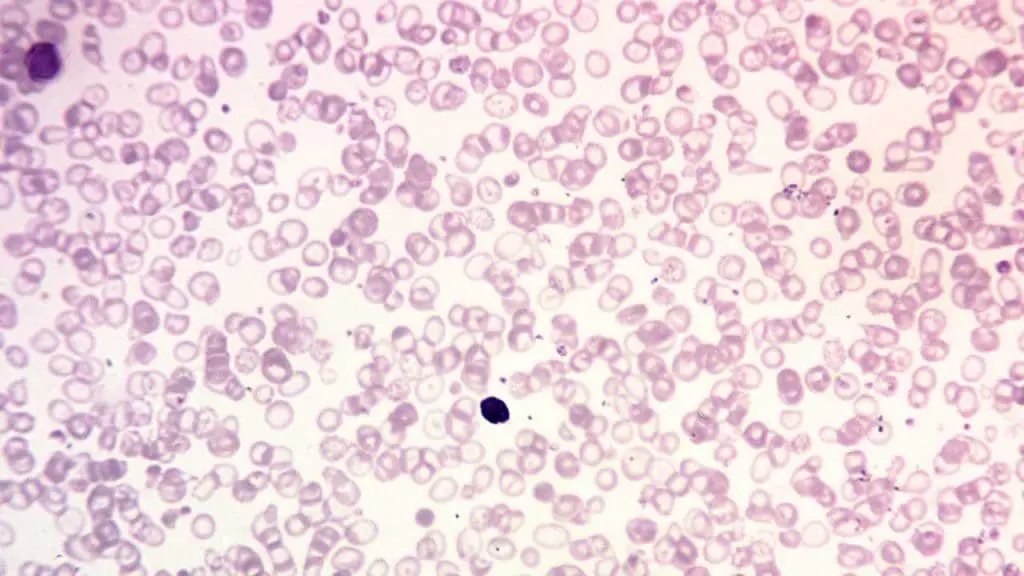
- Peripheral blood smear: To examine red blood cell morphology, which often shows hypochromic, microcytic cells and target cells.
Hemoglobin Studies
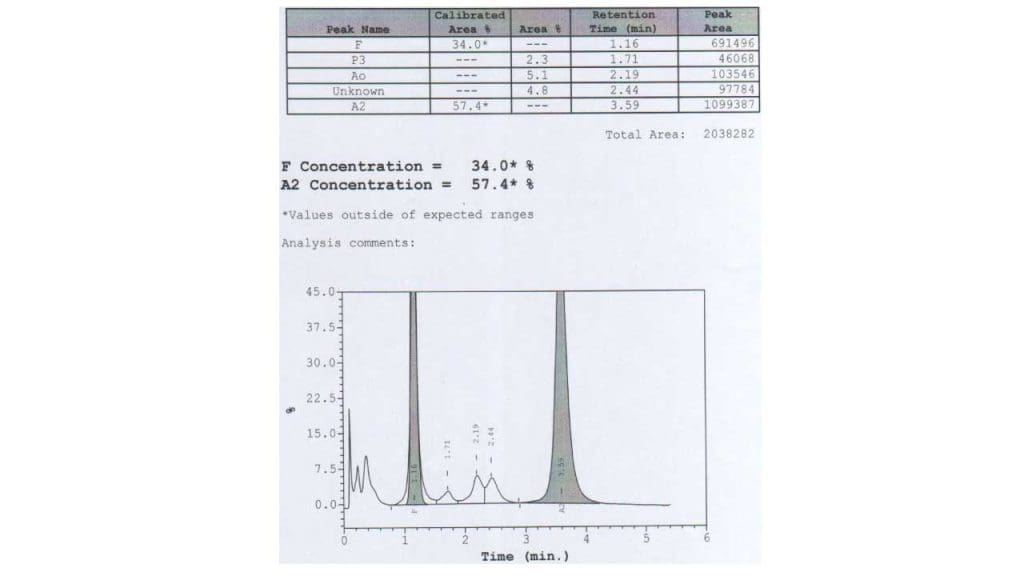
- Hemoglobin electrophoresis or HPLC: To separate different hemoglobin types, essential for diagnosing beta-thalassemia by identifying elevated HbA2 or HbF with reduced or absent HbA. Hb H can be detected on hemoglobin electrophoresis as a fast-moving band
- Hemoglobin A2 measurement: Increased levels of HbA2 are characteristic of beta-thalassemia trait.
- Hemoglobin F measurement: Elevated levels of HbF (fetal hemoglobin) are seen in beta-thalassemia major and intermedia.
Iron Studies
- Serum ferritin: To assess iron stores, which can be normal or elevated in thalassemia, differentiating it from iron deficiency anemia.
- Transferrin saturation: Typically normal or low in thalassemia.
Other Tests
- Genetic testing: Confirmatory test for specific thalassemia mutations.
Differential Diagnosis
It’s essential to differentiate thalassemia from other microcytic anemias, such as iron deficiency anemia and sideroblastic anemia.
- Iron deficiency anemia: Typically has lower ferritin levels, increased transferrin saturation, and different red blood cell morphology (anisocytosis, poikilocytosis).
- Sideroblastic anemia: Characterized by ring sideroblasts in bone marrow, elevated serum iron, and low transferrin saturation.
Treatment and Management
General Principles of Management
The primary goals of thalassemia management are to:
- Improve quality of life.
- Prevent complications.
- Optimize growth and development.
- Prevent morbidity and mortality.
This involves a multidisciplinary approach including hematologists, pediatricians, cardiologists, endocrinologists, and genetic counselors.
Supportive Care
- Regular monitoring: Close follow-up with regular blood tests, including complete blood count, reticulocyte count, and hemoglobin electrophoresis.
- Vaccination: To prevent infections.
- Nutrition: Adequate nutrition is essential for growth and development.
- Education: Patient and family education about the disease, its management, and complications.
Blood Transfusions
- Indication: Used to manage severe anemia in beta-thalassemia major and intermedia, and some cases of hemoglobin H disease.
- Goal: To maintain hemoglobin levels within a target range.
- Frequency: Varies depending on the severity of anemia.
- Complications: Iron overload, transfusion reactions, and alloimmunization.
Iron Chelation Therapy
- Indication: To prevent iron overload caused by frequent blood transfusions.
- Agents: Deferoxamine, deferiprone, and deferasirox.
- Administration: Intravenous, subcutaneous, or oral.
- Goal: To maintain low iron stores to prevent organ damage.
Folic Acid Supplementation
- Indication: To support red blood cell production.
- Dosage: Typically 5 mg daily.
- Importance: Helps in erythropoiesis.
Specific Treatment Options Based on Thalassemia Type
- Alpha-thalassemia: Treatment depends on the severity. Mild forms may require no treatment, while severe forms may require blood transfusions, iron chelation, and potentially bone marrow transplantation.
- Beta-thalassemia: Management focuses on blood transfusions, iron chelation, and supportive care. Bone marrow transplantation is a curative option for selected patients.
Bone Marrow Transplantation
- Indication: Curative treatment for severe forms of thalassemia.
- Requirements: A suitable donor match.
- Procedure: Involves replacing the patient’s bone marrow with healthy donor cells.
- Risks: Graft-versus-host disease, infection, and other transplant-related complications.
Gene Therapy
ZYNTEGLO is a groundbreaking gene therapy developed by Bluebird Bio for the treatment of beta-thalassemia. In addition to ZYNTEGLO, Casgevy (exagamglogene autotemcel) is also approved by the FDA for the treatment of individuals with transfusion-dependent beta thalassemia aged 12 and older.
Both ZYNTEGLO and Casgevy involve modifying the patient’s stem cells with a corrected beta-globin gene and then reintroducing them into the body. While they share similarities, there are differences in the specific gene editing techniques and other aspects of the treatment process.
While they share the common goal of providing a potential cure by modifying the patient’s stem cells with a functional beta-globin gene, they differ in their technological approaches.
ZYNTEGLO
- Vector: Lentiviral vector
- Gene editing: No gene editing
- Mechanism: Integration of a corrected beta-globin gene into the patient’s genome
- Manufacturing: Complex process
Casgevy
- Vector: Self-inactivating lentiviral vector
- Gene editing: CRISPR-Cas9 gene editing
- Mechanism: Precise insertion of corrected beta-globin gene into a specific location
- Manufacturing: Shorter manufacturing timeline
Complications
Thalassemia, while manageable, can lead to a number of complications. These complications often arise from the body’s response to the chronic anemia and ineffective erythropoiesis.
Major Complications
- Iron Overload (Hemochromatosis): This is a common complication, especially in patients requiring frequent blood transfusions. Excess iron can damage the heart, liver, and endocrine glands.
- Bone Problems: The bone marrow expands to compensate for decreased red blood cell production, leading to bone thinning and deformities, especially in the facial bones.
- Splenomegaly: The spleen enlarges due to increased workload in filtering damaged red blood cells. This can lead to discomfort, increased risk of infection, and even rupture.
- Heart Problems: Iron overload and the increased workload on the heart can lead to heart failure, arrhythmias, and other heart complications.
- Liver Problems: Iron overload can damage the liver, leading to cirrhosis and liver failure in severe cases.
- Endocrine Disorders: Iron overload can affect the endocrine system, leading to problems with growth, puberty, and hormone production.
Other Complications
- Growth retardation: Due to chronic anemia and other factors.
- Delayed puberty: Hormonal imbalances can affect puberty.
- Increased risk of infections: Splenectomy can increase susceptibility to infections.
- Osteoporosis: Bone thinning can lead to increased risk of fractures.
- Pulmonary hypertension: In severe cases, due to increased red blood cell breakdown.
It’s important to note that the severity of these complications varies depending on the type of thalassemia and the individual’s response to treatment. Regular monitoring and early intervention are crucial for managing these complications.
Prognosis
The prognosis for thalassemia varies greatly depending on the specific type and severity of the condition.
Mild Forms
- Alpha-thalassemia trait and beta-thalassemia trait typically have a normal life expectancy and minimal symptoms.
- Beta-thalassemia intermedia can have a variable prognosis, but with proper management, many individuals can live normal or near-normal lives.
Severe Forms
- Beta-thalassemia major and hemoglobin H disease were historically associated with a shortened lifespan due to complications from iron overload and organ damage. However, with advancements in medical care, including regular blood transfusions and iron chelation therapy, the prognosis has significantly improved.
- Hydrops fetalis is a severe form of alpha-thalassemia that is usually fatal.
Overall, early diagnosis, consistent medical care, and adherence to treatment plans are crucial for improving the prognosis for individuals with thalassemia.
It’s important to emphasize that while significant progress has been made in managing thalassemia, complications can still arise. Regular monitoring and a multidisciplinary approach to care are essential for optimizing outcomes.
Interaction of Thalassemia with Other Hemoglobinopathies
The interaction of thalassemia with other hemoglobinopathies can lead to complex clinical presentations. Some common examples include:
- Thalassemia and Sickle Cell Disease: Individuals with both thalassemia and sickle cell disease may exhibit a more severe phenotype with increased hemolysis and vaso-occlusive crises.
- Thalassemia and Hemoglobin C Disease: The combination of thalassemia and hemoglobin C disease can lead to a variable clinical course, ranging from mild to severe anemia.
- Thalassemia and Hemoglobin E Disease: This combination is common in Southeast Asian populations and can result in a wide spectrum of clinical severity.
The interaction between thalassemia and other hemoglobinopathies can complicate diagnosis and management. Genetic testing is essential to determine the underlying genetic defects and to guide treatment decisions.
Prevention and Control
Thalassemia is a preventable disease. Effective prevention strategies involve a combination of genetic counseling, prenatal diagnosis, newborn screening, carrier screening, and public health measures.
Genetic Counseling
- Provides information about thalassemia inheritance patterns, risks, and management options.
- Helps couples understand their carrier status and reproductive risks.
- Offers guidance on decision-making regarding family planning.
Prenatal Diagnosis
- Identifies affected fetuses during pregnancy.
- Options include chorionic villus sampling (CVS) and amniocentesis.
- Allows parents to make informed decisions about the pregnancy.
Newborn Screening
- Early detection of thalassemia in newborns.
- Enables timely intervention and management.
- Important for early identification of carriers.
Carrier Screening Programs
- Identify individuals who carry the thalassemia gene.
- Facilitates genetic counseling and reproductive planning.
- Can be offered to specific populations at higher risk.
Public Health Measures
- Awareness campaigns to educate the public about thalassemia.
- Support for thalassemia organizations and patient support groups.
- Development of national thalassemia control programs.
- Collaboration between healthcare providers, policymakers, and communities.
Successful thalassemia prevention programs combine these strategies to reduce the incidence of the disease and improve the quality of life for affected individuals.
It’s important to note that while prenatal diagnosis and termination of affected pregnancies have been effective in reducing the birth of children with severe thalassemia, ethical considerations and cultural sensitivities must be carefully addressed.
Disclaimer: This article is intended for informational purposes only and is specifically targeted towards medical students. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Weatherall D. 2003 William Allan Award address. The Thalassemias: the role of molecular genetics in an evolving global health problem. Am J Hum Genet. 2004 Mar;74(3):385-92. doi: 10.1086/381402. PMID: 15053011; PMCID: PMC1182250.
- Steinberg MH, Forget BG, Higgs DR, Weatherall DJ. Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management (Cambridge Medicine) 2nd Edition. 2009.
- Orkin SH, Nathan DG, Ginsburg D, Look AT, Fisher DE, Samuel Lux MD Nathan and Oski’s Hematology and Oncology of Infancy and Childhood, 2-Volume Set (Saunders) 8th Edition. 2014
- Weatherall D. Thalassaemia: The Biography (Biographies of Disease)(OUP Oxford). 2010.
- https://thalassaemia.org.cy/publications/tif-publications/guidelines-for-the-management-of-%ce%b1-thalassaemia/