Procedure At-A-Glance
HPLC, or high performance liquid chromatography, is the most widely used laboratory method for screening hemoglobinopathies such as beta-thalassemia trait, sickle cell disease, and HbE disease [1,5]. The protocol below describes the Bio-Rad VARIANT II Beta Thalassemia Short Program, a common cation-exchange HPLC system used in clinical labs.
- Collect an EDTA peripheral blood sample.
- Prepare lysate automatically in the sampling station, where the blood is diluted in buffer and red cells are lysed to release hemoglobin.
- Inject the lysate onto a cation-exchange analytical cartridge.
- Separate hemoglobin variants using a phosphate buffer gradient of increasing ionic strength. Each variant elutes at a characteristic retention time.
- Detect at 415 nm with a filter photometer; a 690 nm reading corrects for background.
- Quantify each peak as a percentage of total hemoglobin. The software produces a chromatogram and a report.
- Interpret by matching peak retention times to predefined windows for HbA, HbA2, HbF, HbS, HbC, HbD, and HbE.
Total run time: approximately 6.5 minutes per sample.
Why HPLC Matters in Clinical Practice
Hemoglobinopathies are among the most common single-gene disorders in the world. They include sickle cell disease, alpha- and beta-thalassemias, and structural variants such as HbE, HbC, and HbD. Early identification matters. It guides treatment, supports genetic counseling, and underpins national newborn and antenatal screening programs.
For decades, HPLC has been the routine first-line tool for this work. It is fast, automated, sensitive, and well-suited to high-throughput labs [5,7]. This article walks through how HPLC works, how to read a chromatogram, what its limitations are, and where it sits among newer methods like capillary electrophoresis.
How HPLC Separates Hemoglobin Variants
The principle in one sentence
HPLC separates molecules based on how strongly each one binds to a solid material packed inside a column, while a liquid solvent washes them through.
The stationary phase
The stationary phase is the solid packing inside the column. For hemoglobin analysis, the silica beads are coated with weakly negatively charged groups (typically carboxyl groups). This makes the column a cation-exchange medium. Hemoglobin variants, which carry slightly different positive charges depending on their amino acid sequence, bind to the column with different strengths.
The mobile phase
The mobile phase is the liquid that flows through the column. In Hb HPLC it is a phosphate buffer whose pH and ionic strength are gradually changed during the run. As the salt concentration rises, hemoglobin variants are displaced from the column one by one, in order of how tightly they bind. Stronger binders come off later.
Detection
As each hemoglobin fraction leaves the column, it passes a flow cell where a photometer measures absorbance at 415 nm, the Soret band peak for heme. The software plots absorbance against time, producing the chromatogram: a series of peaks, each corresponding to a hemoglobin variant.
What drives separation
The dominant factor in clinical Hb HPLC is net surface charge. Variants like HbS, HbC, and HbE differ from HbA by single amino acid substitutions that change the protein's charge. The column resolves these differences into distinct peaks. (Hydrophobicity is more important in reversed-phase HPLC, which is used in research settings rather than for routine variant screening.)
Materials
- EDTA peripheral blood sample (or dried blood spot for newborn screening)
- Bio-Rad VARIANT II automated HPLC system
- Diluent buffer
- Two phosphate buffers of different pH and ionic strength
- HbA2/HbF calibrator
- Lyphochek Hemoglobin A2 Control, bilevel (two of each level)
Protocol
- Calibrate. Run the HbA2/HbF calibrator at the start of each batch to generate calibration factors. Run the Lyphochek bilevel control to confirm HbA2 and HbF are within acceptable ranges.
- Inject. Dilute 5 µL of EDTA blood with 1 mL of diluent buffer (handled by the sampling station). Run time: 6.5 minutes per sample.
- Adjust low-Hb samples. For severely anemic samples, manually pre-dilute the whole blood with the kit's wash/diluent solution (typically 1:200, though always verify against the current manufacturer's lot instructions) to ensure the total chromatogram area falls within the valid range of 1,000,000 to 3,000,000 µvolt·second.
- Review each chromatogram against the assigned windows and report HbA, HbA2, HbF, and any abnormal peaks.
Reading a Chromatogram
The HPLC software assigns each peak to a "window" based on its retention time. The table below combines the manufacturer-defined windows with the most common variants found in each window for the Bio-Rad VARIANT II system [4,6].
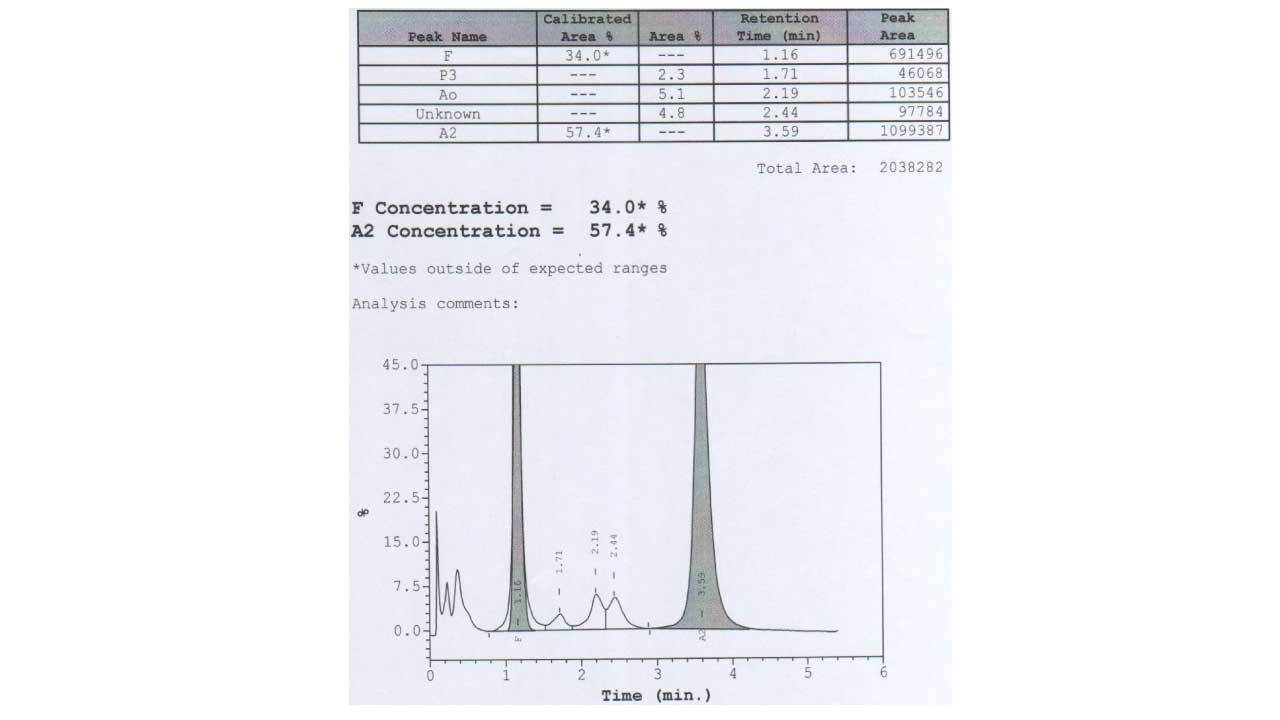
A few interpretation rules students should remember:
- HbA2 above 3.5% in an adult points toward beta-thalassemia trait [6].
- HbA2 above ~10% in the A2 window is more consistent with HbE than true HbA2, because HbE co-elutes with HbA2.
- HbF above 2% in an adult is abnormal and may suggest beta-thalassemia, hereditary persistence of fetal hemoglobin (HPFH), or a recent stress on red cell production.
- An S-window peak around 35–45% in an adult typically indicates sickle cell trait; near 80–90% suggests sickle cell disease.
Worked Example
The chromatogram below shows an elevated HbA2 peak reaching well above the normal 1.5–3.5%. In an adult, an apparent HbA2 of around 20–30% in the A2 window is highly suggestive of homozygous HbE, because HbE co-elutes with HbA2 and the percentage is far too high to be true HbA2. Confirmation by capillary electrophoresis or molecular testing is recommended [1,3].
Limitations and Common Pitfalls
HPLC is reliable, but it is not infallible.
Co-elution. HbE and HbA2 elute in the same window. Hb Lepore, HbD-Iran, and HbG-Coushatta also overlap with HbA2 [3,4]. The percentage helps tell them apart, but molecular confirmation is often needed.
Hb Bart's and HbH. These tetramers of gamma and beta chains, seen in alpha-thalassemia, elute in the pre-integration region (<1 min) and can be missed if the operator does not specifically inspect that area. They are most relevant in newborn screening and in alpha-thalassemia trait [3,5].
Hb Constant Spring. This non-deletional alpha-thalassemia variant, common in Southeast Asia, produces only a small peak in the C-window on HPLC and is easy to overlook. Capillary electrophoresis detects it more reliably [3].
Hb New York and other rare variants. Some variants are simply not separated by HPLC. Comparative studies have shown CE detects certain variants that HPLC misses entirely [2].
Altered HbA2 levels. Coexisting iron deficiency suppresses HbA2 synthesis. A beta-thalassemia trait carrier with iron deficiency may present with a falsely "normal" HbA2 and be missed; best practice is to correct the iron status and repeat [1]. Conversely, severe hyperthyroidism, megaloblastic anemias (B12/folate deficiency), and certain antiretroviral therapies can falsely elevate HbA2, mimicking beta-thalassemia trait [8].
The Borderline Zone. While an HbA2 > 3.5% classically suggests beta-thalassemia trait, modern guidelines caution against treating this as a hard cutoff. Values between 3.3% and 3.7% represent a borderline diagnostic zone. Normal individuals may naturally sit at 3.4%, while patients with "silent" beta-thalassemia mutations may present with an HbA2 of 3.3%. Any result in this borderline zone requires molecular DNA testing for definitive diagnosis [1,8].
Recent transfusion. Donor hemoglobin can dominate the chromatogram for up to 3–4 months after transfusion, masking the patient's true profile.
Sample age and hemolysis. Aged or hemolyzed samples produce spurious peaks from degraded hemoglobin, especially in the P3 window.
HPLC and Capillary Electrophoresis
Capillary electrophoresis (CE) is the main alternative to HPLC for hemoglobinopathy screening. It separates hemoglobin variants by charge-to-size ratio in an electric field rather than on a column.
The two methods agree well for common variants like HbS, HbC, and HbF, but they diverge for some uncommon ones [2]. CE outperforms HPLC for Hb Constant Spring and Hb New York [3]. HPLC tends to give slightly higher HbA2 values than CE, so labs must validate their own cutoffs.
Current guidance treats HPLC and CE as complementary. Many labs use one as the primary screen and the other as the confirmatory method [1,5]. In low-resource settings, an HPLC-first approach has been shown to be the most cost- and time-effective option for thalassemia and common hemoglobinopathy detection [4].
HPLC in Newborn Screening
Newborn screening programs in many countries use HPLC on dried blood spots collected by heel prick. The goal is to identify sickle cell disease and major thalassemias before symptoms appear. Early diagnosis allows prophylactic penicillin in sickle cell disease, appropriate vaccinations, and parental counseling — all of which significantly reduce childhood morbidity and mortality [5,7].
A newborn HPLC pattern is dominated by HbF, with smaller amounts of HbA in healthy babies. The presence of HbS, HbC, or no HbA at all triggers urgent follow-up.
While HPLC and CE remain the standard for adult screening, Tandem Mass Spectrometry (LC-MS/MS) is rapidly becoming the new frontier for high-throughput newborn screening programs. Rather than separating variants by charge, mass spectrometry identifies the exact molecular mass of the mutated globin chains. This allows MS to differentiate variants with identical charges such as HbS, HbC, and HbE with absolute molecular certainty and at incredibly high speeds, making it an increasingly popular choice for national screening programs [7,9].
Frequently Asked Questions (FAQs)
What is HPLC used for in a hematology lab?
HPLC identifies and quantifies hemoglobin variants for the screening of hemoglobinopathies, including beta-thalassemia trait, sickle cell disease, and HbE disease. It is also the most common method for measuring HbA1c in diabetes monitoring.
How long does an HPLC run take?
On legacy systems like the Bio-Rad VARIANT II, a standard run takes about 6.5 minutes per sample. However, modern automated high-throughput systems (such as the Bio-Rad D-100 or Tosoh analyzers) have drastically reduced this time, completing full variant separation in 1 to 3 minutes per sample.
Can HPLC alone diagnose a hemoglobinopathy?
No. HPLC gives a presumptive identification. Current guidelines recommend confirming any significant variant with a second independent method such as capillary electrophoresis, isoelectric focusing, or molecular DNA testing [1].
Why does iron deficiency affect HPLC results?
Iron deficiency can lower HbA2, sometimes pushing it below the 3.5% threshold used to flag beta-thalassemia trait. A patient with both conditions can be falsely reassured. Best practice is to correct the iron deficiency and repeat the HPLC.
Is HPLC reliable for detecting alpha-thalassemia?
Less reliable than for beta-thalassemia. Alpha-thalassemia carriers often have a normal HPLC pattern. Hb Constant Spring, a common non-deletional alpha-thalassemia variant in Southeast Asia, produces only a small C-window peak and is more reliably detected on capillary electrophoresis [3]. Alpha-thalassemia usually requires molecular testing to confirm.
Can HPLC be used on newborns?
Yes. HPLC is a cornerstone of newborn screening programs worldwide. It works on dried blood spots from a heel prick, allowing early detection of sickle cell disease and beta-thalassemia so treatment can start before symptoms appear [5,7].
Glossary of Related Medical Terms
- Chromatogram — the graph produced by HPLC software showing absorbance against time. Each peak is one hemoglobin variant.
- Retention time — the time a hemoglobin variant takes to travel through the column. Each variant has its own characteristic retention time.
- Cation-exchange chromatography — the form of HPLC used for hemoglobin screening; separates molecules by positive charge.
- Hemolysate — a solution containing hemoglobin released from broken-down red blood cells.
- HbA, HbA2, HbF — the three normal hemoglobins. HbA dominates in adults (~96%); HbA2 is minor (~2.5%); HbF is residual (<1%) in adults.
- HbS, HbC, HbE, HbD — the four most common structural variants worldwide.
- Hemoglobinopathy — an inherited disorder of hemoglobin structure or production.
- Beta-thalassemia trait — the carrier state for beta-thalassemia, classically showing HbA2 above 3.5% on HPLC.
- Co-elution — when two different variants leave the column at nearly the same time, making them hard to distinguish.
- Capillary electrophoresis (CE) — an alternative method that separates hemoglobins by charge in an electric field; often used to confirm HPLC findings.
Disclaimer: This protocol is for educational purposes only. Local laboratory standard operating procedures take precedence. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. Always consult a qualified healthcare professional for clinical decision-making. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Bain, B. J., Daniel, Y., Henthorn, J., de la Salle, B., Hogan, A., Roy, N. B. A., Mooney, C., Langabeer, L., Rees, D. C., & BSH Committee (2023). Significant haemoglobinopathies: A guideline for screening and diagnosis: A British Society for Haematology Guideline: A British Society for Haematology Guideline. British journal of haematology, 201(6), 1047–1065. https://doi.org/10.1111/bjh.18794
- Asghar, S., Mahmood Abbasi, A., Zafar, J., Mahmood, R., Zahir, S., & Khan, M. (2024). Comparison of High-Performance Liquid Chromatography and Capillary Electrophoresis in the Screening of Haemoglobinopathies. Pakistan Armed Forces Medical Journal, 74(2), 357-361. https://doi.org/10.51253/pafmj.v74i2.7452
- Embong, S. F., Daud, A., Nordin, M. H., & Adzahar, S. (2024). Detection of Hemoglobin Constant Spring: A Comparison of Capillary Electrophoresis Versus High-Performance Liquid Chromatography. Cureus, 16(8), e67228. https://doi.org/10.7759/cureus.67228
- Periyavan S, Kumar S, Mamatha GN, Hegde S, Jain S, Dhanya R, Agarwal RK and Faulkner L (2025) HPLC first approach in detecting thalassemia and other common hemoglobinopathies is more cost and time effective. Front. Hematol. 4:1461498. https://doi.org/10.3389/frhem.2025.1461498
- Frömmel C. (2018). Newborn Screening for Sickle Cell Disease and Other Hemoglobinopathies: A Short Review on Classical Laboratory Methods-Isoelectric Focusing, HPLC, and Capillary Electrophoresis. International journal of neonatal screening, 4(4), 39. https://doi.org/10.3390/ijns4040039
- Khera, R., Singh, T., Khuana, N., Gupta, N., & Dubey, A. P. (2015). HPLC in characterization of hemoglobin profile in thalassemia syndromes and hemoglobinopathies: a clinicohematological correlation. Indian journal of hematology & blood transfusion : an official journal of Indian Society of Hematology and Blood Transfusion, 31(1), 110–115. https://doi.org/10.1007/s12288-014-0409-x
- Zhang, C., Chen, V. C., Osa-Andrews, B., & Cao, J. (2025). Emerging Technologies and Advanced Strategies in Hemoglobin Defect Screening. Journal of clinical medicine, 14(16), 5690. https://doi.org/10.3390/jcm14165690
- Mosca, A., Paleari, R., Ivaldi, G., Galanello, R., & Giordano, P. C. (2009). The role of haemoglobin A(2) testing in the diagnosis of thalassaemias and related haemoglobinopathies. Journal of clinical pathology, 62(1), 13–17. https://doi.org/10.1136/jcp.2008.056945
- Daniel, Y. A., Turner, C., Haynes, R. M., Hunt, B. J., & Dalton, R. N. (2005). Rapid and specific detection of clinically significant haemoglobinopathies using electrospray mass spectrometry-mass spectrometry. British journal of haematology, 130(4), 635–643. https://doi.org/10.1111/j.1365-2141.2005.05646.x