Key Takeaways
Sideroblastic anemia is a group of inherited and acquired disorders in which bone marrow cells cannot build iron into hemoglobin, leaving iron stranded inside red cell precursors as ring sideroblasts visible on Prussian blue staining [3,9].
- Causes ▾: Inherited forms most often involve ALAS2 mutations (X-linked sideroblastic anemia), while acquired forms include MDS with ring sideroblasts and reversible triggers like alcohol, lead, isoniazid, linezolid, and copper deficiency [1,2,10].
- Signs and Symptoms ▾: The clinical presentation of sideroblastic anemia consists of anemia-related symptoms like fatigue, pallor, and shortness of breath, coupled with multi-organ complications from iron overload such as hepatosplenomegaly, cardiac arrhythmias, liver damage, and endocrine dysfunction [3,6].
- Laboratory Investigations ▾: Diagnosis requires at least 15% ring sideroblasts among bone marrow erythroblasts, or at least 5% when an SF3B1 mutation is confirmed by molecular testing [4,6,7].
- Treatment and Management ▾: Treatment is cause-specific: high-dose vitamin B6 for many ALAS2 cases, luspatercept as first-line therapy for transfusion-dependent lower-risk MDS-RS, imetelstat for ESA-refractory cases (FDA-approved June 2024), and iron chelation for transfusional iron overload [12,13,14]. The Molecular International Prognostic Scoring System (IPSS-M), introduced in 2022, integrates 31 gene mutations with clinical features to give the most precise risk estimate currently available for MDS [15].
*Click ▾ for more information
Introduction
Sideroblastic anemia is not one disease but a family of disorders that share a single mechanical fault: red blood cell precursors in the bone marrow cannot build iron into hemoglobin. Iron arrives at the cell as expected. The cell takes it in. But somewhere in the eight-step process of heme synthesis, the assembly line breaks down. Iron accumulates inside mitochondria — the cell's energy factories — and forms a blue ring around the nucleus when stained with Prussian blue dye [3,9].
These distinctive cells are called ring sideroblasts. Their presence in the bone marrow defines sideroblastic anemia. Diagnosis requires at least 15% of erythroblasts to be ring sideroblasts, or at least 5% if a specific gene mutation called SF3B1 is detected [4,6,7].
The name is misleading in one important way. Despite being a disease of iron, sideroblastic anemia is rarely caused by a lack of iron. The body usually has plenty. The problem is that cells cannot use what they have. The result is a paradox: the bone marrow is iron-rich, yet hemoglobin-poor.
Sideroblastic anemia is uncommon. Inherited forms are rare (fewer than one case per million for X-linked disease). The most clinically important acquired form, MDS with ring sideroblasts (MDS-RS), accounts for roughly 10–15% of all myelodysplastic syndromes and typically presents in adults over 60 [2,4].
The Role of Heme Synthesis
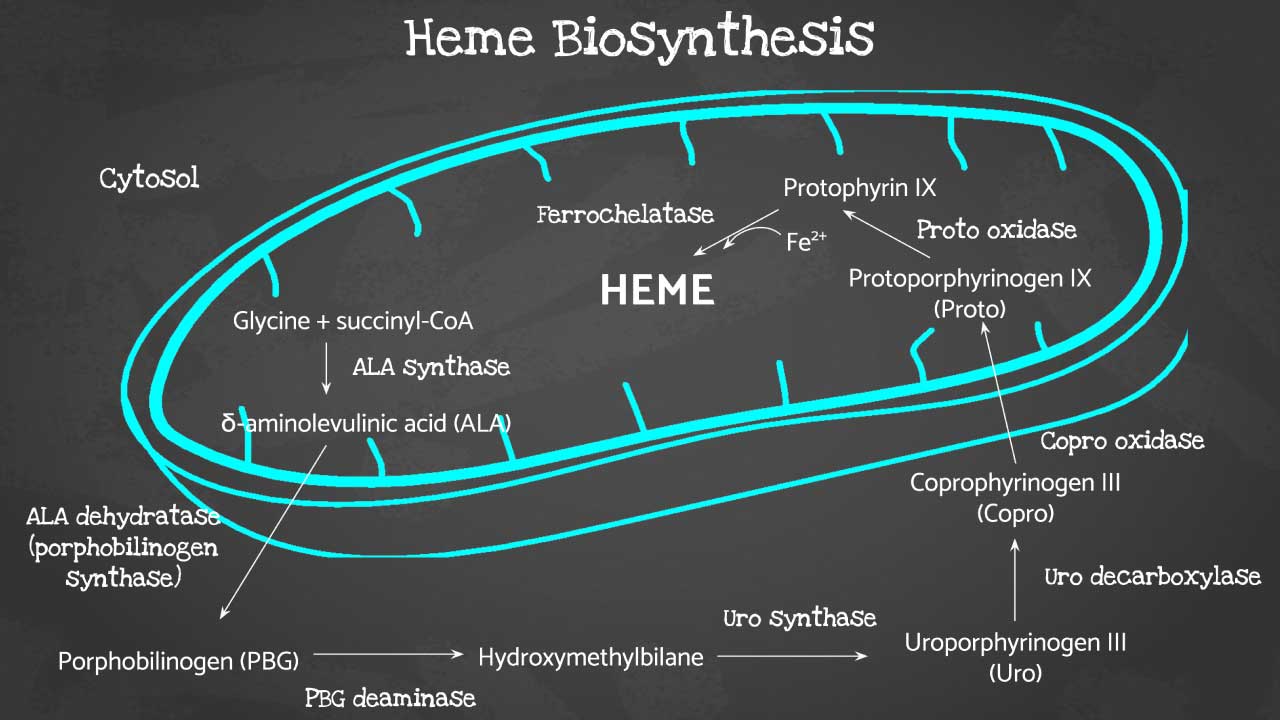
Heme is the iron-containing core of hemoglobin. Without heme, hemoglobin cannot carry oxygen. The body builds heme through an eight-step chemical pathway that starts in the mitochondria, moves out into the cytosol (the fluid inside the cell), and returns to the mitochondria to finish [1,9].
The first step is rate-limiting. An enzyme called ALAS2 (aminolevulinic acid synthase 2) combines two molecules, glycine and succinyl-CoA, to make ALA (5-aminolevulinic acid). ALAS2 needs vitamin B6 (pyridoxine) as a helper molecule [1,11]. Without ALAS2 — or without B6 — the entire pathway stalls before it begins.
The final step matters too. An enzyme called ferrochelatase inserts iron into protoporphyrin IX to produce heme [1,11]. Lead poisoning blocks ferrochelatase, which is why lead exposure causes sideroblastic anemia.
When any step in this chain fails, iron still arrives at the mitochondria but cannot be locked into heme. It collects there because mitochondria are where iron handling occurs. Over time, the iron-laden mitochondria pile up around the nucleus of the erythroblast and form the ring sideroblast [9].
Causes of Sideroblastic Anemia
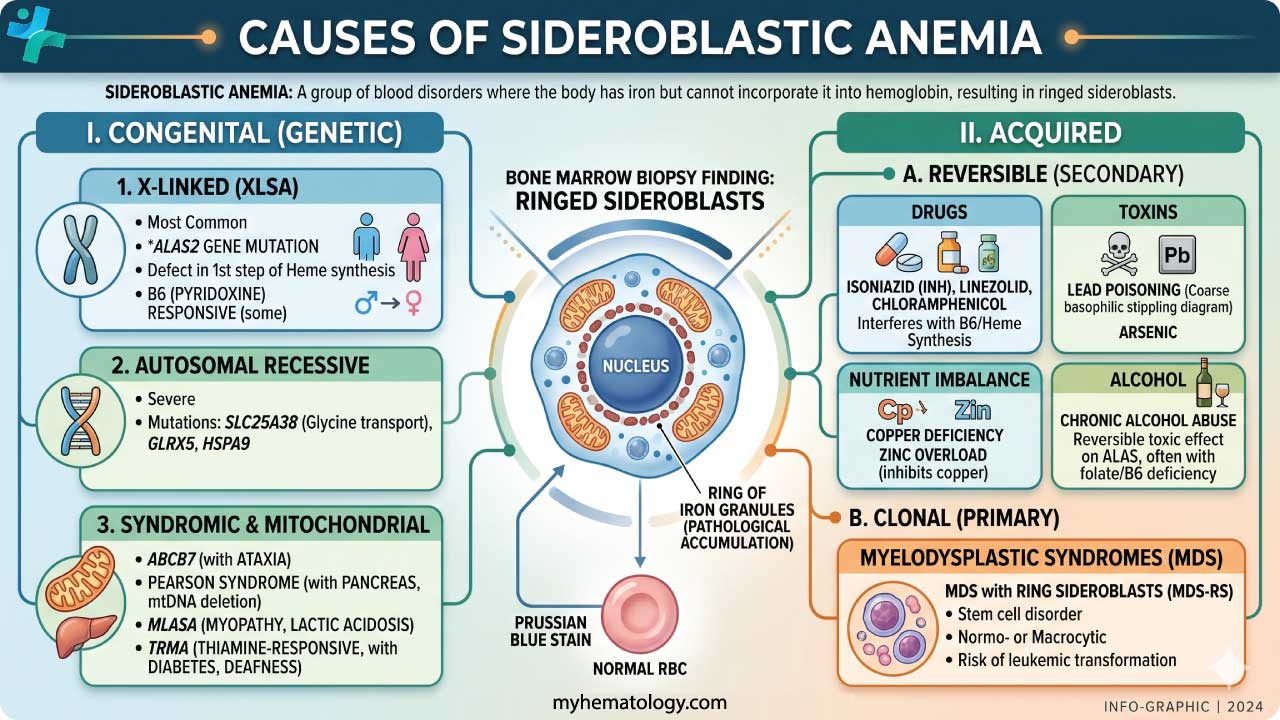
Sideroblastic anemia is divided into two broad groups: inherited (congenital) forms and acquired forms. The biology underneath each differs, but the end result is the same — ring sideroblasts and ineffective erythropoiesis (red blood cells that are made but die before leaving the marrow) [2,3].
Inherited Sideroblastic Anemias
Inherited forms results from gene mutations affecting proteins involved in heme synthesis or mitochondrial iron handling. They typically appear in childhood or early adulthood and are lifelong conditions [8,9].
X-linked Sideroblastic Anemia (XLSA)
XLSA is the most common inherited form. It is caused by mutations in ALAS2 on the X chromosome. Males, who have only one X chromosome, are typically affected. Female carriers usually have mild or no symptoms because of X-inactivation, the process in which one X chromosome is randomly switched off in each cell [1,11]. Many patients with XLSA respond at least partially to high-dose vitamin B6, which stabilizes the ALAS2 enzyme. The response depends on the exact mutation, and hemoglobin rarely returns fully to normal [10,11].
A separate, rarer condition — XLSA with cerebellar ataxia (XLSA/A) — is caused by ABCB7 mutations and includes poor balance and coordination. It is a distinct diagnosis [1].
Autosomal Recessive Sideroblastic Anemias (ARSA)
ARSA is a group of conditions caused by mutations in various genes involved in heme synthesis and mitochrondrial function [1,8,9]:
- SLC25A38 encodes a mitochondrial transporter that delivers glycine to ALAS2. Without it, ALAS2 has no substrate.
- GLRX5 encodes glutaredoxin 5, which helps assemble iron-sulfur clusters inside mitochondria.
- HSPA9 encodes a mitochondrial chaperone protein involved in iron-sulfur cluster formation.
- TRNT1 mutations cause SIFD syndrome, combining sideroblastic anemia with immunodeficiency, fevers, and developmental delay.
Mitochondrial Sideroblastic Anemias
A distinct subgroup is caused by deletions or mutations in mitochondrial DNA. The most important example is Pearson marrow-pancreas syndrome, a serious condition that typically presents in infancy. It causes transfusion-dependent anemia alongside failure of the exocrine pancrease. Large mitochondrial DNA deletions are responsible, and the prognosis is poor [1].
Acquired Sideroblastic Anemias
Acquired forms develop later in life. Identifying the trigger matters because many acquired forms are at least partly reversible [2,10].
MDS with Ring Sideroblasts (MDS-RS) and SF3B1 Mutations
The most clinically important acquired cause is a myelodysplastic syndrome with ring sideroblasts (MDS-RS). MDS is a clonal bone marrow disorder, meaning it grows from a single abnormal stem cell that copies itself. MDS-RS predominantly affects older adults, with median age around 70 [4,5].
Around 80–90% of MDS-RS cases are driven by mutations in SF3B1, a gene that encodes part of the spliceosome — the cellular machinery that cuts and joins RNA so genes can be read correctly. When SF3B1 is mutated, key transcripts including those for mitochondrial iron handling proteins are spliced incorrectly, which impairs heme synthesis and produces ring sideroblasts [4,5].
When an SF3B1 mutation is detected, the diagnostic threshold for ring sideroblasts drops from 15% to 5%. The mutation also confirms the disease is clonal and predicts a relatively favorable prognosis compared with other MDS subtypes [4,7].
Under the 2022 WHO 5th edition and the 2022 International Consensus Classification, the older label Refractory Anemia with Ring Sideroblasts has been retired. The current entity is MDS with low blasts and SF3B1 mutation (MDS-SF3B1), with morphologically defined subtypes available for cases without SF3B1 mutations [6,7]. This change reflects a conceptual shift: the genetic lesion now defines the entity, not the morphology alone.
Alcohol-Related Sideroblastic Anemia
CChronic alcohol use disrupts red blood cell production through several mechanisms running at once [2,10]:
- Direct toxicity to bone marrow cells.
- Damage to mitochondria inside erythroblasts.
- Suppression of hepcidin (the liver hormone that controls iron absorption), which leads to excess iron uptake from the gut.
- Depletion of vitamin B6, folate, and copper.
- Increased oxidative stress from alcohol metabolism.
This form is often substantially reversible with sustained abstinence and nutritional rehabilitation [10].
Medications
Several drugs can trigger sideroblastic anemia. Isoniazid and ethionamide (used to treat tuberculosis) interfere with vitamin B6 metabolism. Linezolid (an antibiotic) and chloramphenicol damage mitochondria directly. In all these cases, stopping the offending drug is the first move [2,10].
Heavy Metal Toxicity
Lead poisoning blocks several enzymes in the heme pathway, including ALA dehydratase and ferrochelatase. Arsenic can also disrupt red cell production. Both deserve consideration when patients have occupational or environmental exposure histories [3,10].
Nutritional Deficiencies
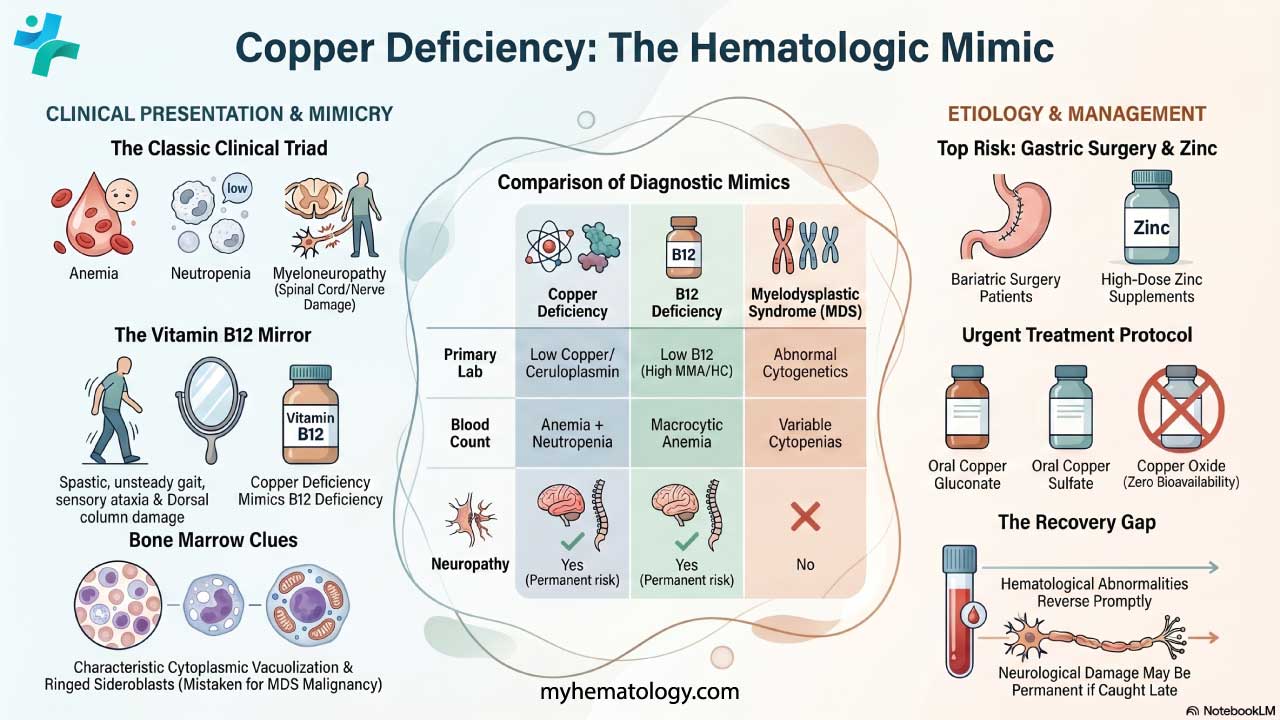
Vitamin B6 deficiency impairs ALAS2 activity and often contributes alongside alcohol use or malnutrition. Copper deficiency is an underappreciated cause, particularly in patients on long-term intravenous (parenteral) nutrition or after bariatric (weight-loss) surgery, where copper absorption is reduced [10,19].
When to Suspect an Inherited Cause
A few clinical patterns should prompt genetic workup [1,8,9]:
- Onset in childhood or early adulthood.
- A family history of unexplained anemia.
- Severe microcytic anemia that does not respond to iron.
- Failure to respond to obvious reversible triggers.
Signs and Symptoms
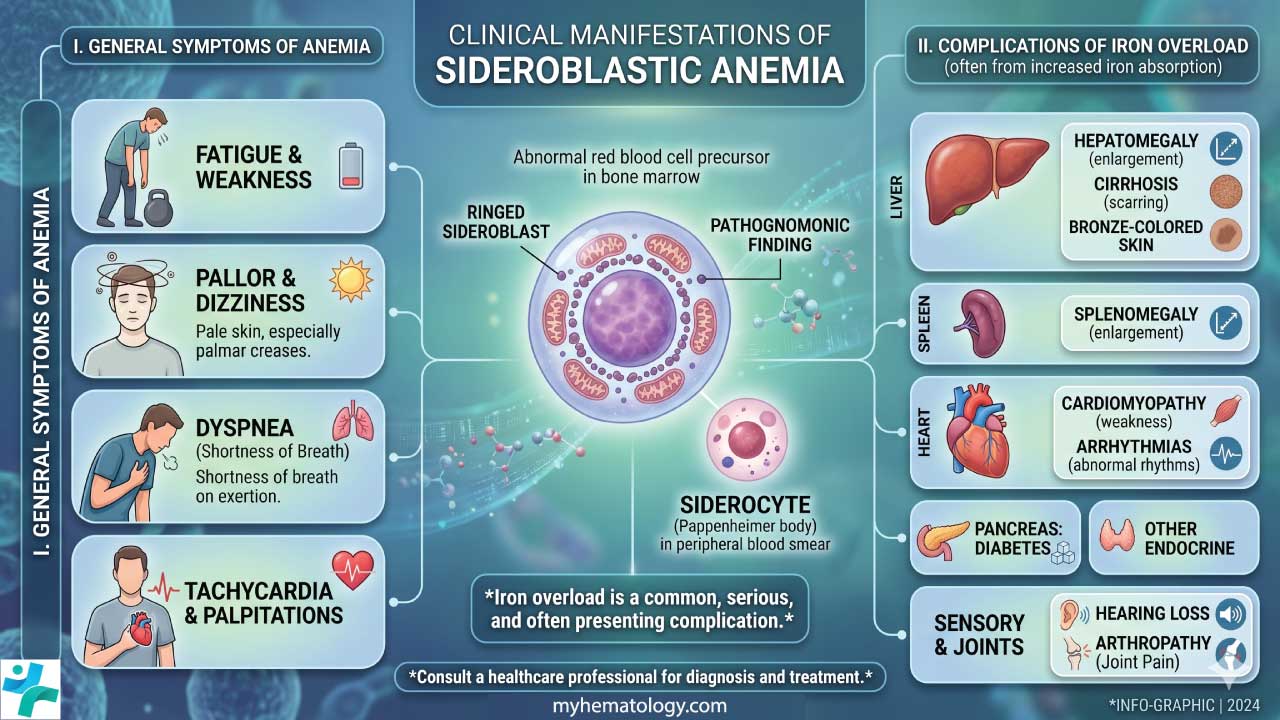
Sideroblastic anemia produces symptoms from two processes running in parallel. The anemia itself reduces oxygen delivery. The iron that cannot be used builds up in organs over time [3,16]. How prominent each is depends on the subtype, severity, and how long the patient has had the condition.
Anemia Symptoms
These reflect low hemoglobin and reduced oxygen delivery to tissues [2]:
- Fatigue and generalized weakness, usually the most prominent complaint.
- Pallor of the skin and inner eyelids (conjunctivae), reflecting low circulating hemoglobin.
- Shortness of breath on exertion.
- Palpitations and tachycardia, as the heart works harder to compensate.
- Dizziness or light-headedness, particularly on standing.
- Reduced exercise tolerance.
Iron Overload Features
When iron cannot enter hemoglobin, it deposits in the liver, heart, endocrine glands, and joints. Transfusion-dependent patients accumulate iron faster because each unit of red cells carries roughly 200–250 mg of iron [16,17]:
- Hepatomegaly and, eventually, liver fibrosis or cirrhosis.
- Cardiomyopathy or arrhythmias from cardiac iron deposition.
- Diabetes mellitus and endocrine dysfunction from pancreatic and pituitary involvement.
- Skin bronzing in long-standing iron overload.
- Joint pain from iron in synovial tissue.
Other Signs
- Splenomegaly may occur from extramedullary hematopoiesis (blood production outside the bone marrow) or from increased destruction of abnormal red cells [2].
- Developmental delay and pancreatic insufficiency in infants point toward Pearson syndrome [1].
- Signs of chronic liver disease or peripheral neuropathy often coexist in alcohol-related forms [10].
Laboratory Investigations
Diagnosing sideroblastic anemia requires a combination of blood tests, bone marrow examination, and — in selected cases — molecular genetic testing. Bone marrow examination with Prussian blue staining remains the gold standard [3,4].
Complete Blood Count (CBC) and Reticulocyte Count
- Anemia pattern: Microcytic and hypochromic in inherited XLSA. Acquired forms, especially alcohol-related, are often macrocytic [10,11].
- MCV: Low in microcytic forms, normal or high in macrocytic acquired forms [10].
- Dimorphic red cell population: Two distinct populations — one normal-sized, one small and pale — is a characteristic clue, especially in XLSA and after pyridoxine therapy [9].
- Hemoglobin and hematocrit: Both reduced.
- Reticulocyte count: Normal or inappropriately low for the degree of anemia, reflecting ineffective erythropoiesis rather than blood loss or hemolysis.
Peripheral Blood Smear
Careful smear examination can reveal red blood cell abnormalities like [3,9,10]:
- Target cells and hypochromic cells in inherited forms.
- A dimorphic red cell population.
- Anisocytosis and poikilocytosis (variable size and shape of red cells).
- Basophilic stippling — small blue dots inside red cells, most prominent in lead-related sideroblastic anemia.
Bone Marrow Examination with Prussian Blue Stain

This is the definitive test. A bone marrow aspirate is stained with Prussian blue, which binds selectively to non-heme iron. Ring sideroblasts are erythroblasts in which iron-laden mitochondria form a perinuclear ring encircling at least one-third of the nucleus. Normal sideroblasts contain only a few scattered iron granules [3,4].
The diagnostic thresholds are:
- At least 15% ring sideroblasts among nucleated erythroblasts, or
- At least 5% ring sideroblasts when an SF3B1 mutation is confirmed [4,6,7].
The bone marrow examination also assesses erythroid hyperplasia, the degree of dysplasia (especially in MDS-RS), and overall marrow architecture [3,9].
Iron Studies
Iron studies in sideroblastic anemia contrast sharply with iron deficiency anemia [2,16]:
- Serum iron: Normal or elevated. Iron is available but not used.
- Serum ferritin: Normal or elevated. Stores are full or overflowing.
- Transferrin saturation: Often elevated, sometimes above 45%.
- TIBC: Normal or low.
Sideroblastic Anemia vs Iron Deficiency Anemia
This distinction matters clinically. Treating sideroblastic anemia with iron supplements can cause real harm.
| Feature | Iron Deficiency Anaemia | Sideroblastic Anaemia |
| Serum iron | Low | Normal, elevated, or variable |
| Serum ferritin | Very low | Normal or elevated |
| TIBC | High | Normal or low |
| Transferrin saturation | Low (<16%) | Elevated (often >45%) |
| MCV | Microcytic | Microcytic or macrocytic |
| Blood film | Microcytic, hypochromic | Dimorphic population; basophilic stippling if lead-related |
| Bone marrow iron stores | Absent or depleted | Normal or increased |
| Ring sideroblasts | Absent | ≥15% (or ≥5% if SF3B1 mutated) |
| Response to iron supplements | Yes | No — may worsen iron overload |
Additional and Molecular Tests
- Genetic testing: Recommended in young patients or those with a family history; panels covering ALAS2, SLC25A38, GLRX5, HSPA9, and mitochondrial DNA deletions [1,8].
- Next-Generation Sequencing (NGS): Recommended for all adults with acquired ring sideroblasts. Beyond confirming SF3B1 mutations, NGS provides the data needed to calculate the IPSS-M score, the current gold-standard prognostic tool. IPSS-M integrates clinical variables with mutations in 31 genes and sorts patients into six risk categories: Very Low, Low, Moderate-Low, Moderate-High, High, and Very High [4,15].
- Blood lead level: If lead poisoning is suspected based on history or basophilic stippling [10].
- Serum copper and ceruloplasmin: To exclude copper deficiency, particularly in patients on parenteral nutrition or following bariatric surgery [19].
- Serum vitamin B6: In patients with alcohol dependence or malnutrition [10].
Differential Diagnosis
Several conditions can mimic sideroblastic anemia and should be excluded before making the diagnosis [2,3]:
- Iron deficiency anemia: Low serum ferritin and transferrin saturation — the opposite of sideroblastic anemia [20].
- Thalassemia trait: Microcytic anemia with normal or elevated iron studies; diagnosed by hemoglobin electrophoresis or molecular testing [10].
- Anemia of chronic disease: Low serum iron but elevated ferritin; no ring sideroblasts [16].
- Lead poisoning: Causes sideroblastic anemia with prominent basophilic stippling; confirmed by blood lead measurement [10].
- Congenital dyserythropoietic anemias (CDAs): Ineffective erythropoiesis with distinct bone marrow morphology and specific genetic causes [1].
- Other MDS subtypes: May have ring sideroblasts but a more complex clinical picture and worse prognosis [6,7].
Treatment and Management of Sideroblastic Anemia
Treatment depends on the underlying cause, the severity of anemia, and whether iron overload or other complications are present. The goals are to relieve symptoms, prevent organ damage from accumulated iron, and, where possible, address the root cause [2,3].
Addressing the Underlying Cause
For acquired forms, identifying and removing the trigger is the first step [10]:
- Alcohol: Sustained abstinence combined with nutritional rehabilitation (B6, folate, copper) can result in significant improvement within weeks to months.
- Medications: Stopping the offending drug and supplementing pyridoxine typically leads to resolution.
- Heavy metal toxicity: Chelation specific to the metal (e.g., EDTA for lead) and removal from the source.
- Nutritional deficiencies: Vitamin B6, copper, or folate supplementation as indicated.
Pyridoxine (Vitamin B6) Therapy for XLSA
High-dose pyridoxine (typically 50–200 mg daily) is the first-line treatment for X-linked sideroblastic anemia. It stabilizes ALAS2 and partially restores heme synthesis. The response depends on the specific mutation. Pyridoxine does not correct the underlying genetic defect, so long-term monitoring of blood counts and iron studies remains essential [1,10,11].
Supportive Therapy
- Red blood cell transfusions: Indicated for symptomatic severe anemia. Each unit of packed red cells delivers approximately 200-250 mg of iron, so transfusion-dependent patients inevitably accumulate iron overload over time. Transfusion thresholds should be individualised [16,17].
- Folic acid supplementation: Recommended in all patients, particularly when folate deficiency coexists, to support erythropoiesis.
- Erythropoiesis-stimulating agents (ESAs): Erythropoietin or darbepoetin can reduce transfusion needs in lower-risk MDS-RS patients, particularly those with low baseline serum erythropoietin. Their frontline role has been challenged by newer targeted agents [12,13].
Luspatercerpt - An Emerging Standard of Care for MDS-RS
Luspatercept is a targeted drug that helps red blood cells finish maturing. It works by blocking abnormal TGF-β signaling that suppresses erythroblast development. Originally approved by the FDA in 2020 for patients who had not responded to ESAs, its indication was expanded in 2023 to cover first-line treatment for ESA-naive adults with transfusion-dependent lower-risk MDS. The phase 3 COMMANDS trial showed luspatercept achieved superior transfusion independence and longer-lasting hemoglobin gains compared with standard ESA therapy [12,13].
Response rates are highest in patients with SF3B1 mutations, which makes luspatercept especially well-suited to MDS-RS. It is given as a subcutaneous injection every three weeks and is generally well-tolerated [13].
Imetelstat - A Novel Telomerase Inhibitor
Imetelstat received FDA approval in June 2024 for adults with lower-risk MDS and transfusion-dependent anemia who have not responded to, have lost response to, or are unsuitable for ESAs. It blocks telomerase — the enzyme that abnormal bone marrow clones rely on to keep dividing. Rather than treating symptoms alone, imetelstat targets the underlying clone biology. The phase 3 IMerge trial demonstrated meaningful transfusion independence in a previously hard-to-treat population. It is given as an intravenous infusion every four weeks. The main toxicities are thrombocytopenia and neutropenia, requiring careful blood count monitoring [14].
Hypomethylating Agents
Azacitidine and decitabine are reserved for higher-risk MDS rather than lower-risk MDS-RS, where they offer little advantage over targeted agents. They exist in the broader MDS treatment landscape but are not first-line for sideroblastic anemia.
Iron Chelation Therapy (for Iron Overload)
Iron overload is a major cause of morbidity and mortality in patients receiving regular transfusions. Chelation Iron overload is a major source of long-term morbidity and mortality in transfusion-dependent patients. Chelation binds excess iron so it can be excreted [17]:
- Deferoxamine is given as a subcutaneous or intravenous infusion, usually overnight. It is highly effective but inconvenient.
- Deferasirox is taken once daily as an oral tablet. It is generally first-line for long-term chelation. Renal function and urinary protein should be monitored.
- Deferiprone is an oral agent with particular efficacy against cardiac iron. It may be combined with deferoxamine in severe cardiac iron overload.
Chelation is typically initiated when serum ferritin consistently exceeds 1,000 µg/L in transfusion-dependent patients, or when end-organ iron deposition is detected by MRI. Cardiac and hepatic T2* MRI sequences are the gold standard for monitoring [17].
Allogeneic Stem Cell Transplantation
Allogeneic hematopoietic stem cell transplant remains the only potentially curative option for severe inherited sideroblastic anemias and for selected MDS-RS cases. Because of significant transplant-related morbidity and mortality, it is reserved for [9,10]:
- Children or young adults with severe transfusion-dependent inherited forms (particularly autosomal recessive types) who have a suitable matched donor.
- Higher-risk MDS-RS patients who have progressed or are ineligible for other therapies.
General and Long-Term Management
- Regular monitoring includes complete blood counts, iron studies, and assessment for end-organ damage [3].
- Dietary guidance: Patients with iron overload should avoid iron-fortified supplements and avoid taking high-dose vitamin C with meals (vitamin C enhances iron absorption). Alcohol should be moderated [16,17].
- Genetic counseling is essential for patients with inherited forms and their families [10].
- Psychosocial support: Chronic anemia and ongoing transfusions affect quality of life. Access to mental health support and patient communities matters more than it is sometimes credited for.
Overall Prognosis
Prognosis varies widely by subtype. Acquired reversible forms generally have excellent outcomes once the trigger is removed. Inherited forms are lifelong but are typically compatible with a good quality of life under modern management.
For MDS-RS, IPSS-M is now the definitive prognostic tool. While SF3B1 mutations on their own predict a favorable course, IPSS-M accounts for co-mutations that can shift the picture. This molecular scoring helps clinicians identify patients who may benefit from earlier intervention with targeted therapies or transplantation [4,15].
Frequently Asked Questions (FAQs)
What is sideroblastic anemia in simple terms?
Sideroblastic anemia is a group of blood disorders in which the bone marrow cannot use iron properly to make hemoglobin. Iron is available, but red blood cell precursors fail to build it into the oxygen-carrying part of red cells. Instead, iron piles up in the mitochondria of these cells, forming a distinctive blue ring under the microscope when stained with Prussian blue. The result is anemia together with, over time, iron overload affecting the liver, heart, and other organs [3,4].
Should patients with sideroblastic anemia take iron supplements?
No. This is one of the most important points for patients and caregivers to understand. In sideroblastic anemia, the body usually has enough or even too much iron — the problem is that cells cannot use it. Iron supplements can worsen iron overload in the liver, heart, and endocrine glands. This is the opposite of iron deficiency anemia, where supplements are part of treatment. Anyone diagnosed with sideroblastic anemia should check with their hematologist before taking any iron-containing vitamin or supplement [2,16].
Is sideroblastic anemia hereditary?
Some forms are, but most are not. Inherited forms, such as X-linked sideroblastic anemia from ALAS2 mutations, run in families and usually appear in childhood or early adulthood. Acquired forms develop later in life from causes such as alcohol use, certain medications (isoniazid, linezolid), heavy metal exposure, copper deficiency, or a bone marrow disorder called MDS with ring sideroblasts. A family history, young age at diagnosis, or severe microcytic anemia that does not respond to treatment all raise suspicion for an inherited cause [1,8].
Why does vitamin B6 deficiency cause sideroblastic anemia?
Vitamin B6 (pyridoxine) is the helper molecule for ALAS2, the enzyme that starts heme synthesis. ALAS2 needs B6 to combine glycine and succinyl-CoA into ALA, the first building block of heme. When B6 is deficient, ALAS2 activity drops and ALA supply falls. The whole heme pathway stalls. Iron still arrives at the mitochondria of red cell precursors, but it cannot be built into heme, so it accumulates and forms ring sideroblasts [1,11].
Why does copper deficiency cause sideroblastic anemia?
Copper is a cofactor for several enzymes important to red blood cell production, including cytochrome c oxidase in the mitochondrial electron transport chain and hephaestin, which helps the body absorb iron from the diet. When copper is low, mitochondrial energy production suffers, heme synthesis becomes inefficient, and iron handling inside erythroblasts is disrupted. The combination produces ring sideroblasts and anemia. Copper deficiency should be considered in patients on long-term intravenous nutrition or after bariatric surgery [10,19].
What is a ring sideroblast and why does it matter?
A ring sideroblast is an immature red blood cell (erythroblast) that has failed to incorporate iron into hemoglobin. Instead, iron piles up inside the cell's mitochondria and, when stained with Prussian blue, appears as a ring of blue granules around the nucleus. Diagnostic thresholds require at least 15% ring sideroblasts among erythroblasts, or at least 5% when an SF3B1 mutation is confirmed. Their presence reflects a specific problem with iron use inside red cell precursors — not a shortage of iron in the body [3,4,7].
Can sideroblastic anemia be cured?
It depends on the cause. Acquired forms from alcohol, medications, or vitamin deficiencies often improve substantially or fully resolve once the trigger is removed and nutrition is restored. Inherited forms are lifelong but can be managed well with pyridoxine, iron chelation, and regular monitoring. For severe inherited cases in young patients, allogeneic stem cell transplantation offers a potential cure. MDS-RS is not usually curable with standard therapy, but newer drugs like luspatercept and imetelstat have meaningfully improved transfusion independence and quality of life [10,12,13,14].
What is iron overload and how serious is it?
Iron overload occurs when excess iron accumulates in body tissues — particularly the liver, heart, and endocrine glands. In sideroblastic anemia, iron builds up both because the normal process of incorporation into hemoglobin is disrupted and because each transfused unit of blood adds approximately 200-250 mg of iron. Over time, this causes liver fibrosis or cirrhosis, cardiac arrhythmias, diabetes, and hormonal problems. It is monitored using serum ferritin and MRI T2* sequences, and managed with chelation therapy when ferritin consistently exceeds 1,000 µg/L [16,17].
What is SF3B1 and why is it important?
SF3B1 encodes a component of the spliceosome — the cellular machinery that processes RNA. Mutations in SF3B1 are found in roughly 80–90% of patients with MDS-RS. These mutations disrupt normal RNA processing in erythroblasts, impairing mitochondrial function and heme synthesis. Testing for SF3B1 matters for three reasons: it confirms the process is clonal, it lowers the diagnostic threshold for ring sideroblasts from 15% to 5%, and SF3B1-mutant MDS-RS carries a more favorable prognosis and predicts a stronger response to luspatercept [4,5,7].
What is luspatercept and is it widely available?
Luspatercept is a targeted drug that promotes red blood cell maturation by blocking TGF-β signalling pathways. While originally approved by the FDA in 2020 specifically for patients who had not responded to erythropoiesis-stimulating agents (ESAs), subsequent studies (such as the phase 3 COMMANDS trial) demonstrated its superiority over standard ESAs. As a result, it is now approved as a frontline (first-line) treatment for adults with transfusion-dependent lower-risk MDS. It is administered by subcutaneous injection every three weeks. Availability varies by country and health system; patients should discuss eligibility with their hematologist [13].
Glossary of Related Medical Terms
- ALAS2 (Aminolevulinic Acid Synthase 2): The enzyme catalysing the first step of heme synthesis in erythroblasts. Its gene is located on the X chromosome; loss-of-function mutations cause XLSA.
- Allogeneic Stem Cell Transplantation: A procedure in which a patient receives blood-forming stem cells from a matched donor, potentially replacing diseased bone marrow with healthy donor cells. The only curative option for severe inherited sideroblastic anemia.
- Anemia: A reduction in red blood cells or hemoglobin below normal levels, impairing oxygen delivery to tissues.
- Basophilic Stippling: Bluish granules within red blood cells on a peripheral blood smear, representing aggregated ribosomes. Prominently associated with lead poisoning-related sideroblastic anemia.
- Bone Marrow: Spongy tissue inside bones responsible for producing blood cells. In sideroblastic anaemia, the marrow produces abnormal erythroblasts that fail to mature into healthy red blood cells.
- Deferasirox: An oral iron chelating agent taken once daily, used to reduce iron overload in patients receiving regular blood transfusions.
- Deferoxamine: An iron chelating agent administered by subcutaneous or intravenous infusion. One of the oldest and most established treatments for transfusion-related iron overload.
- Dimorphic Red Cell Population: A peripheral blood film finding in which two distinct populations of red cells are visible — one normal-sized and one microcytic/hypochromic. Characteristic of sideroblastic anemia.
- Erythroblast (Red Blood Cell Precursor): An immature cell in the bone marrow that develops into a red blood cell. In sideroblastic anemia, erythroblasts accumulate abnormal iron deposits and fail to mature.
- Erythropoiesis: The process by which new red blood cells are produced in the bone marrow. 'Ineffective erythropoiesis' occurs when erythroblasts are produced in large numbers but fail before reaching the circulation.
- Ferrochelatase: The enzyme catalysing the final step of heme synthesis — inserting iron into protoporphyrin IX. Its inhibition by lead contributes to lead-related sideroblastic anemia.
- Ferritin: A protein that stores iron within cells. Serum ferritin is a marker of total body iron stores — elevated in iron overload; reduced in iron deficiency.
- GLRX5: A gene encoding glutaredoxin 5, involved in mitochondrial iron-sulphur cluster assembly. Mutations cause an autosomal recessive form of sideroblastic anemia.
- Hemoglobin: The iron-containing protein inside red blood cells that binds and transports oxygen. Each hemoglobin molecule contains four heme units; disrupted heme synthesis leads to hemoglobin deficiency.
- Heme: The iron-containing component of hemoglobin, synthesised through an eight-step enzymatic pathway. Disruption of any step causes iron to accumulate rather than be incorporated into hemoglobin.
- Hepcidin: A hormone produced by the liver that regulates iron absorption from the gut. Alcohol suppresses hepcidin, leading to excess iron absorption.
- Imetelstat: A first-in-class telomerase inhibitor approved in 2024 for adults with lower-risk, transfusion-dependent MDS. It targets the underlying biology of malignant clones and is used for patients who are ineligible for or have stopped responding to erythropoiesis-stimulating agents (ESAs).
- Iron Chelation Therapy: Treatment using drugs that bind to excess iron in the body and allow it to be excreted, preventing accumulation and organ damage.
- Iron Overload (Secondary Haemosiderosis): Excess iron deposition in tissues and organs including the liver, heart, and endocrine glands. A major long-term complication of sideroblastic anemia.
- Luspatercept: A recombinant fusion protein that promotes red blood cell maturation by reducing aberrant TGF-β signalling. Originally approved in 2020 for ESA-refractory patients, it is now an established frontline (first-line) therapy for ESA-naïve adults with transfusion-dependent lower-risk MDS.
- MCV (Mean Corpuscular Volume): A measurement of the average size of red blood cells. Low in most inherited forms; normal or elevated in some acquired forms.
- Myelodysplastic Syndrome with Ring Sideroblasts (MDS-RS): A clonal bone marrow disorder predominantly affecting older adults, caused in most cases by SF3B1 mutations. Carries a relatively favorable prognosis among MDS subtypes.
- Pearson Marrow-Pancreas Syndrome: A rare mitochondrial disorder of infancy caused by large mitochondrial DNA deletions, presenting with severe sideroblastic anemia and exocrine pancreatic insufficiency.
- Prussian Blue Stain (Perls’ Stain): A chemical stain applied to bone marrow aspirates to detect non-heme iron deposits, revealing the characteristic blue perinuclear ring in sideroblastic anemia.
- Pyridoxine (Vitamin B6): A B-vitamin that acts as a cofactor for ALAS2. High-dose supplementation is the first-line treatment for XLSA and can partially restore heme synthesis.
- Ring Sideroblast: An erythroblast with iron-laden mitochondria arranged in a perinuclear ring visible on Prussian blue staining, occupying at least one-third of the nuclear circumference. The hallmark finding in sideroblastic anemia.
- SF3B1: A gene encoding a core component of the RNA spliceosome. Somatic mutations found in approximately 80-90% of MDS-RS cases, lowering the diagnostic ring sideroblast threshold from 15% to 5%.
- SLC25A38: A gene encoding a mitochondrial glycine transporter. Mutations cause a common form of autosomal recessive sideroblastic anemia.
- Splenomegaly: Abnormal enlargement of the spleen, resulting from extramedullary hematopoiesis or increased destruction of abnormal red blood cells.
- Transferrin Saturation: A blood test measuring the proportion of iron-transport protein (transferrin) currently carrying iron. Often elevated in sideroblastic anemia.
- X-Linked Sideroblastic Anaemia (XLSA): The most common inherited form of sideroblastic anemia, caused by ALAS2 mutations on the X chromosome. Predominantly affects males and is often partially responsive to pyridoxine supplementation.
Disclaimer: This article is intended for informational purposes only and is specifically targeted towards medical students. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Ducamp, S., & Fleming, M. D. (2019). The molecular genetics of sideroblastic anemia. Blood, 133(1), 59–69. https://doi.org/10.1182/blood-2018-08-815951
- Abu-Zeinah, G., & DeSancho, M. T. (2020). Understanding Sideroblastic Anemia: An Overview of Genetics, Epidemiology, Pathophysiology and Current Therapeutic Options. Journal of blood medicine, 11, 305–318. https://doi.org/10.2147/JBM.S232644
- Bottomley, S. S., & Fleming, M. D. (2014). Sideroblastic anemia: diagnosis and management. Hematology/oncology clinics of North America, 28(4), 653–v. https://doi.org/10.1016/j.hoc.2014.04.008
- Malcovati, L., Karimi, M., Papaemmanuil, E., Ambaglio, I., Jädersten, M., Jansson, M., Elena, C., Gallì, A., Walldin, G., Della Porta, M. G., Raaschou-Jensen, K., Travaglino, E., Kallenbach, K., Pietra, D., Ljungström, V., Conte, S., Boveri, E., Invernizzi, R., Rosenquist, R., Campbell, P. J., … Hellström Lindberg, E. (2015). SF3B1 mutation identifies a distinct subset of myelodysplastic syndrome with ring sideroblasts. Blood, 126(2), 233–241. https://doi.org/10.1182/blood-2015-03-633537
- Papaemmanuil, E., Cazzola, M., Boultwood, J., Malcovati, L., Vyas, P., Bowen, D., Pellagatti, A., Wainscoat, J. S., Hellstrom-Lindberg, E., Gambacorti-Passerini, C., Godfrey, A. L., Rapado, I., Cvejic, A., Rance, R., McGee, C., Ellis, P., Mudie, L. J., Stephens, P. J., McLaren, S., Massie, C. E., … Chronic Myeloid Disorders Working Group of the International Cancer Genome Consortium (2011). Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. The New England journal of medicine, 365(15), 1384–1395. https://doi.org/10.1056/NEJMoa1103283
- Arber, D. A., Orazi, A., Hasserjian, R. P., Borowitz, M. J., Calvo, K. R., Kvasnicka, H. M., Wang, S. A., Bagg, A., Barbui, T., Branford, S., Bueso-Ramos, C. E., Cortes, J. E., Dal Cin, P., DiNardo, C. D., Dombret, H., Duncavage, E. J., Ebert, B. L., Estey, E. H., Facchetti, F., Foucar, K., … Tefferi, A. (2022). International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood, 140(11), 1200–1228. https://doi.org/10.1182/blood.2022015850
- Khoury, J. D., Solary, E., Abla, O., Akkari, Y., Alaggio, R., Apperley, J. F., Bejar, R., Berti, E., Busque, L., Chan, J. K. C., Chen, W., Chen, X., Chng, W. J., Choi, J. K., Colmenero, I., Coupland, S. E., Cross, N. C. P., De Jong, D., Elghetany, M. T., Takahashi, E., … Hochhaus, A. (2022). The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia, 36(7), 1703–1719. https://doi.org/10.1038/s41375-022-01613-1
- Bergmann, A. K., Campagna, D. R., McLoughlin, E. M., Agarwal, S., Fleming, M. D., Bottomley, S. S., & Neufeld, E. J. (2010). Systematic molecular genetic analysis of congenital sideroblastic anemia: evidence for genetic heterogeneity and identification of novel mutations. Pediatric blood & cancer, 54(2), 273–278. https://doi.org/10.1002/pbc.22244
- Fleming M. D. (2011). Congenital sideroblastic anemias: iron and heme lost in mitochondrial translation. Hematology. American Society of Hematology. Education Program, 2011, 525–531. https://doi.org/10.1182/asheducation-2011.1.525
- Alcindor, T., & Bridges, K. R. (2002). Sideroblastic anaemias. British journal of haematology, 116(4), 733–743. https://doi.org/10.1046/j.0007-1048.2002.03378.x
- Cox, T. C., Bottomley, S. S., Wiley, J. S., Bawden, M. J., Matthews, C. S., & May, B. K. (1994). X-linked pyridoxine-responsive sideroblastic anemia due to a Thr388-to-Ser substitution in erythroid 5-aminolevulinate synthase. The New England journal of medicine, 330(10), 675–679. https://doi.org/10.1056/NEJM199403103301004
- Fenaux, P., Platzbecker, U., Mufti, G. J., Garcia-Manero, G., Buckstein, R., Santini, V., Díez-Campelo, M., Finelli, C., Cazzola, M., Ilhan, O., Sekeres, M. A., Falantes, J. F., Arrizabalaga, B., Salvi, F., Giai, V., Vyas, P., Bowen, D., Selleslag, D., DeZern, A. E., Jurcic, J. G., … List, A. F. (2020). Luspatercept in Patients with Lower-Risk Myelodysplastic Syndromes. The New England journal of medicine, 382(2), 140–151. https://doi.org/10.1056/NEJMoa1908892
- Platzbecker, U., Della Porta, M. G., Santini, V., et al. (2023). Efficacy and safety of luspatercept versus epoetin alfa in erythropoiesis-stimulating agent-naive, transfusion-dependent, lower-risk myelodysplastic syndromes (COMMANDS): interim analysis of a phase 3, open-label, randomised controlled trial. The Lancet, 402(10399), 373–385. https://doi.org/10.1016/S0140-6736(23)00874-7
- Platzbecker, U., Santini, V., Fenaux, P., et al. (2024). Imetelstat in patients with lower-risk myelodysplastic syndromes who have relapsed or are refractory to erythropoiesis-stimulating agents (IMerge): a multinational, randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet, 403(10423), 249–260. https://doi.org/10.1016/S0140-6736(23)01724-5
- Bernard, E., Tuechler, H., Greenberg, P. L., et al. (2022). Molecular International Prognostic Scoring System for Myelodysplastic Syndromes. NEJM Evidence, 1(7). https://doi.org/10.1056/EVIDoa2200008
- Coates T. D. (2014). Physiology and pathophysiology of iron in hemoglobin-associated diseases. Free radical biology & medicine, 72, 23–40. https://doi.org/10.1016/j.freeradbiomed.2014.03.039
- Gattermann N. (2008). Overview of guidelines on iron chelation therapy in patients with myelodysplastic syndromes and transfusional iron overload. International journal of hematology, 88(1), 24–29. https://doi.org/10.1007/s12185-008-0118-z
- Kaur A, Chhabra A. Sideroblastic Anemia. [Updated 2024 Dec 11]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2026 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK538287/
- Jaiser, S. R., & Winston, G. P. (2010). Copper deficiency myelopathy. Journal of neurology, 257(6), 869–881. https://doi.org/10.1007/s00415-010-5511-x
- Goddard, A. F., James, M. W., McIntyre, A. S., Scott, B. B., & British Society of Gastroenterology (2011). Guidelines for the management of iron deficiency anaemia. Gut, 60(10), 1309–1316. https://doi.org/10.1136/gut.2010.228874
- Cazzola, M., Invernizzi, R., Bergamaschi, G., Levi, S., Corsi, B., Travaglino, E., Rolandi, V., Biasiotto, G., Drysdale, J., & Arosio, P. (2003). Mitochondrial ferritin expression in erythroid cells from patients with sideroblastic anemia. Blood, 101(5), 1996–2000. https://doi.org/10.1182/blood-2002-07-2006
- Camaschella C. (2009). Hereditary sideroblastic anemias: pathophysiology, diagnosis, and treatment. Seminars in hematology, 46(4), 371–377. https://doi.org/10.1053/j.seminhematol.2009.07.001
- Steensma D. P. (2015). Myelodysplastic Syndromes: Diagnosis and Treatment. Mayo Clinic proceedings, 90(7), 969–983. https://doi.org/10.1016/j.mayocp.2015.04.001
- Cazzola, M., Malcovati, L., & Invernizzi, R. (2011). Myelodysplastic/myeloproliferative neoplasms. Hematology. American Society of Hematology. Education Program, 2011, 264–272. https://doi.org/10.1182/asheducation-2011.1.264
- Pearson, H. A., Lobel, J. S., Kocoshis, S. A., Naiman, J. L., Windmiller, J., Lammi, A. T., Hoffman, R., & Marsh, J. C. (1979). A new syndrome of refractory sideroblastic anemia with vacuolization of marrow precursors and exocrine pancreatic dysfunction. The Journal of pediatrics, 95(6), 976–984. https://doi.org/10.1016/s0022-3476(79)80286-3
- Lill, R., Hoffmann, B., Molik, S., Pierik, A. J., Rietzschel, N., Stehling, O., Uzarska, M. A., Webert, H., Wilbrecht, C., & Mühlenhoff, U. (2012). The role of mitochondria in cellular iron-sulfur protein biogenesis and iron metabolism. Biochimica et biophysica acta, 1823(9), 1491–1508. https://doi.org/10.1016/j.bbamcr.2012.05.009
- Heeney, M. M., & Finberg, K. E. (2014). Iron-refractory iron deficiency anemia (IRIDA). Hematology/oncology clinics of North America, 28(4), 637–v. https://doi.org/10.1016/j.hoc.2014.04.009
- Platzbecker, U., Germing, U., Götze, K. S., Kiewe, P., Mayer, K., Chromik, J., Radsak, M., Wolff, T., Zhang, X., Laadem, A., Sherman, M. L., Attie, K. M., & Giagounidis, A. (2017). Luspatercept for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes (PACE-MDS): a multicentre, open-label phase 2 dose-finding study with long-term extension study. The Lancet. Oncology, 18(10), 1338–1347. https://doi.org/10.1016/S1470-2045(17)30615-0