Procedure-at-a-Glance
A PCR protocol for the beta-globin (HBB) gene amplifies the entire 1.6 kb gene so it can be sequenced and screened for mutations that cause beta thalassemia.
- Thaw reagents and UV-sterilize the workstation.
- Make a master mix by combining polymerase, dNTPs, buffers and primers.
- Add ~300 ng patient DNA.
- Amplify the sequences using a thermal cycler.
- Check products on a 1.5% agarose gel.
- Purify products enzymatically or with a column.
- Send for Sanger sequencing.
What is Beta Thalassemia?
Beta thalassemia is an inherited blood disorder caused by mutations in the HBB gene, which codes for beta-globin chains in the hemoglobin tetramer [1,2]. When beta-globin is reduced or absent, red blood cells become small and pale, and patients develop anemia of varying severity. The clinical spectrum ranges from beta thalassemia minor (carrier state, mild or no symptoms) to transfusion-dependent thalassemia, which requires lifelong management [5].
More than 350 different HBB mutations have been identified [7]. Most are point mutations or small insertions and deletions inside the gene or its regulatory regions. Identifying the exact mutation matters for prognosis, genetic counseling, prenatal diagnosis, and increasingly for treatment decisions in the era of gene therapy.
HBB Gene
Where It Sits
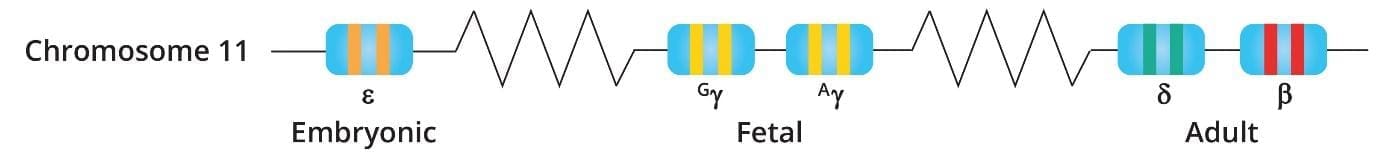
The HBB gene sits on the short (p) arm of chromosome 11 at position 15.4. It is part of the beta-globin gene cluster, which also contains genes for fetal and embryonic hemoglobin chains.
Structure
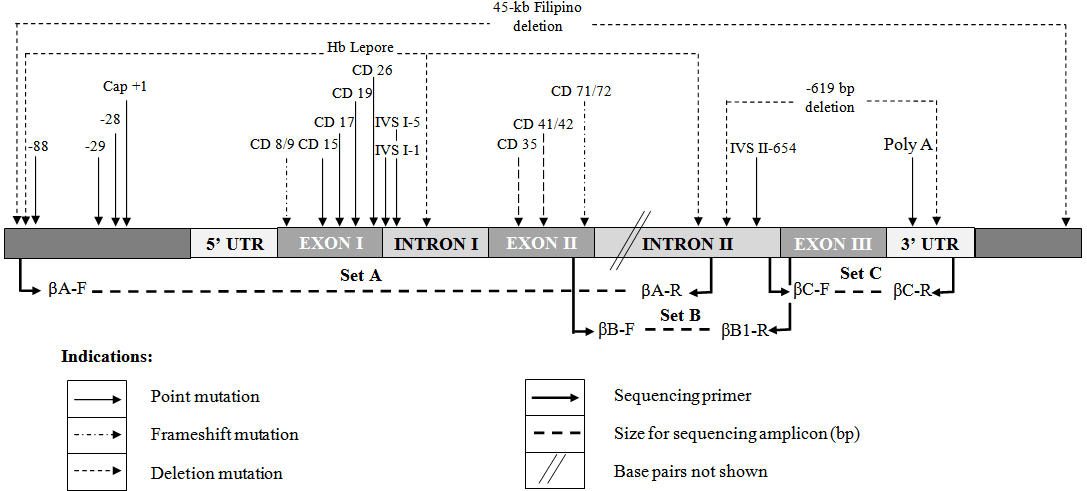
The gene is small but layered. It contains three exons (coding regions), two introns (non-coding regions), a promoter that controls expression, and untranslated regions at both ends. Mutations can occur anywhere along this stretch. Some of the most common beta thalassemia mutations sit inside introns — IVS1-5 and IVS2-654 are well-known examples — which is why a sequencing strategy must cover the entire gene, not just the exons [3,4].
Why Sequence the Whole Gene?
Targeted assays like ARMS-PCR only check a short list of known mutations. In a cosmopolitan city with lots of immigrants, there are just too many mutations to cover using ARMS-PCR only. If a patient carries a rare or novel variant, those tests will also miss it. Direct sequencing of the full 1.6 kb of HBB is the way to catch any variant in the amplified region, including ones not seen before in that population [4].
Principle of Conventional PCR Protocol
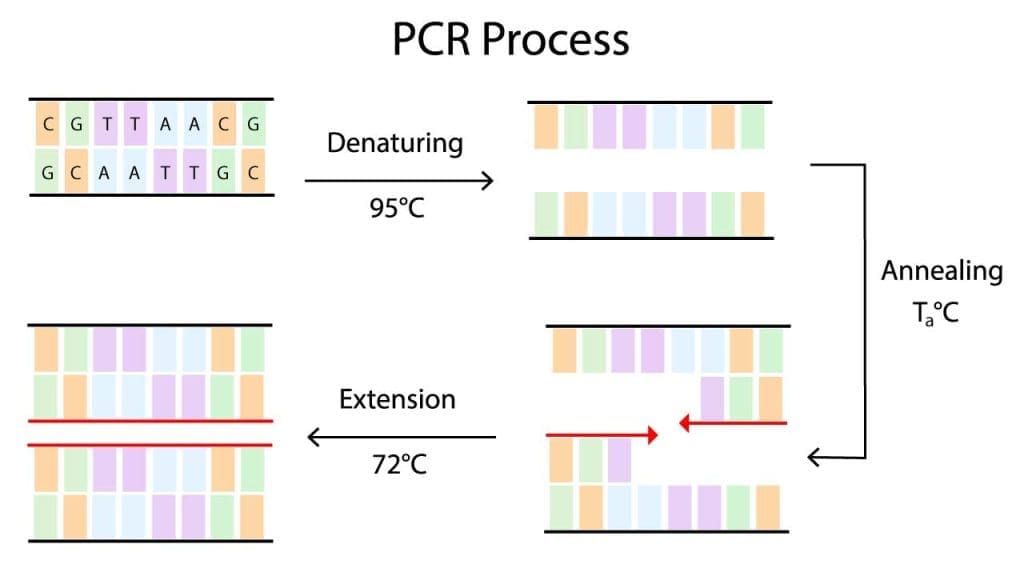
Polymerase chain reaction (PCR) amplifies a small piece of DNA into millions of identical copies. Each cycle has three steps:
Denaturation. The reaction is heated to about 95°C. Hydrogen bonds between the two DNA strands break, separating them.
Annealing. The temperature drops to a primer-specific value (the annealing temperature, or Ta is usually between 55°C and 70°C). Short DNA primers bind to their matching sequences on the template.
Extension. The temperature rises to about 72°C. A heat-stable DNA polymerase builds a new strand starting from each primer, using the template as a guide.
These three steps repeat 30 times in this protocol. Each cycle roughly doubles the amount of target DNA, so 30 cycles produce around a billion copies of the original sequence. That gives enough material for downstream sequencing.
For sequencing applications, modern molecular diagnostics labs use high-fidelity polymerases (such as NEB Q5 or Invitrogen Platinum SuperFi) instead of standard Taq. These enzymes possess 3′→5′ exonuclease ("proofreading") activity, resulting in error rates up to 100 times lower than standard Taq [8]. This is critical because a polymerase-induced mistake during amplification will carry over into Sanger sequencing and can be misread as a pathogenic patient variant.
Materials
Reagents
- PCR master mix (containing polymerase, dNTPs, buffer, and Mg²⁺) — high-fidelity polymerase recommended for sequencing applications
- Forward and reverse primers (10 µM stock, see Table 1 for sequences)
- Nuclease-free water
- Genomic DNA template (~100 ng/µL)
- 70% ethanol or isopropanol for surface decontamination
Consumables
- Filter (aerosol-barrier) pipette tips
- 1.5 mL microcentrifuge tubes
- 0.2 mL PCR tubes or 8-well strips
- Disposable gloves
Equipment
- Calibrated micropipettes (range covering 0.5 µL to 1000 µL)
- Vortex mixer
- Microcentrifuge
- Thermal cycler
- UV-equipped PCR workstation or laminar flow cabinet
PCR Protocol
Primer Design
The protocol uses three primer pairs to cover the HBB gene in three overlapping segments. Sequences and conditions are shown in Table 1 [10].
| Primer ID | Sequence (5′ → 3′) | Position (HUMHBB U01317) | Ta (°C) | Amplicon (bp) | |
|---|---|---|---|---|---|
| βA-F FWD | CGA TCT TCA ATA TGC TTA CCA A | 61830–61851 | 60 | 916 | |
| βA-R REV | CAT TCG TCT GTT TCC CAT TCT A | 62745–62725 | 60 | 916 | |
| βB-F FWD | GCA CGT GGA TCC TGA GAA CT | 62607–62626 | 68 | 902 | |
| βB-R REV | CAC ACA GAC CAG CAC GTT G | 63508–63490 | 68 | 902 | |
| βC-F FWD | GCT AAT CAT GTT CAT ACC TCT T | 63445–63466 | 62 | 855 | |
| βC-R REV | CAG ATT CCG GGT CAC TGT G | 64299–64281 | 62 | 855 | |
Pre-PCR Setup
Before any reagent is opened, contamination control is the priority. Beta thalassemia diagnostic labs typically follow the EMQN best-practice approach [4]: separate pre-PCR and post-PCR rooms, dedicated pipettes for each area, and aliquoted reagents to limit how often a master tube is opened.
- Wipe down the bench, pipettes, and tube racks with 70% alcohol. Switch on the UV lamp in the PCR workstation for at least 10 minutes.
- Thaw reagents and primer aliquots on ice, vortex briefly, then spin down to collect at the bottom of each tube.
Master Mix Preparation
Prepare the master mix on ice in a 1.5 mL microcentrifuge tube. Always make slightly more than you need (typically n+1 reactions) to account for pipetting losses.
| Component | Volume / reaction | Final concentration |
|---|---|---|
| 2× PCR master mix | 25 µL | 1× |
| Forward + reverse primer mix (10 µM each) | 4 µL | 0.4 µM each |
| Nuclease-free water | 18 µL | — |
| Subtotal (master mix) | 47 µL | — |
| Template DNA (~100 ng/µL) | 3 µL | ~300 ng total |
| Final reaction volume | 50 µL | — |
Setting Up the Reaction
- Mix the master mix gently and spin down briefly.
- Aliquot 47 µL of master mix into each PCR tube, kept on ice.
- Add 3 µL of template DNA per sample. Include one no-template control per primer set, replacing template with nuclease-free water.
- Cap, vortex briefly, and spin down.
Thermal Cycling
- Load tubes into the thermal cycler and run the program in Table 3, using the Ta value matched to the primer set.
| Step | Temperature | Time |
|---|---|---|
|
Initial Denaturation
|
98°C | 30 s - 3 min |
|
30×
Denaturation
|
98°C | 10 s |
|
30×
Annealing
|
Ta
60, 68, or 62°C
|
30 s |
|
30×
Extension
|
72°C | 30 s |
|
Final Extension
|
72°C | 2 min |
|
Hold
|
4°C | ∞ |
High-fidelity polymerases typically require a higher denaturation temperature (98°C) than standard Taq (95°C) and have much faster extension rates. Always verify specific temperature and activation time requirements with the manufacturer's insert.
- After cycling, run products on a 1–1.5% agarose gel to verify amplification (see Interpretation).
- Purify successful PCR products before sending for Sanger sequencing.
Interpretation
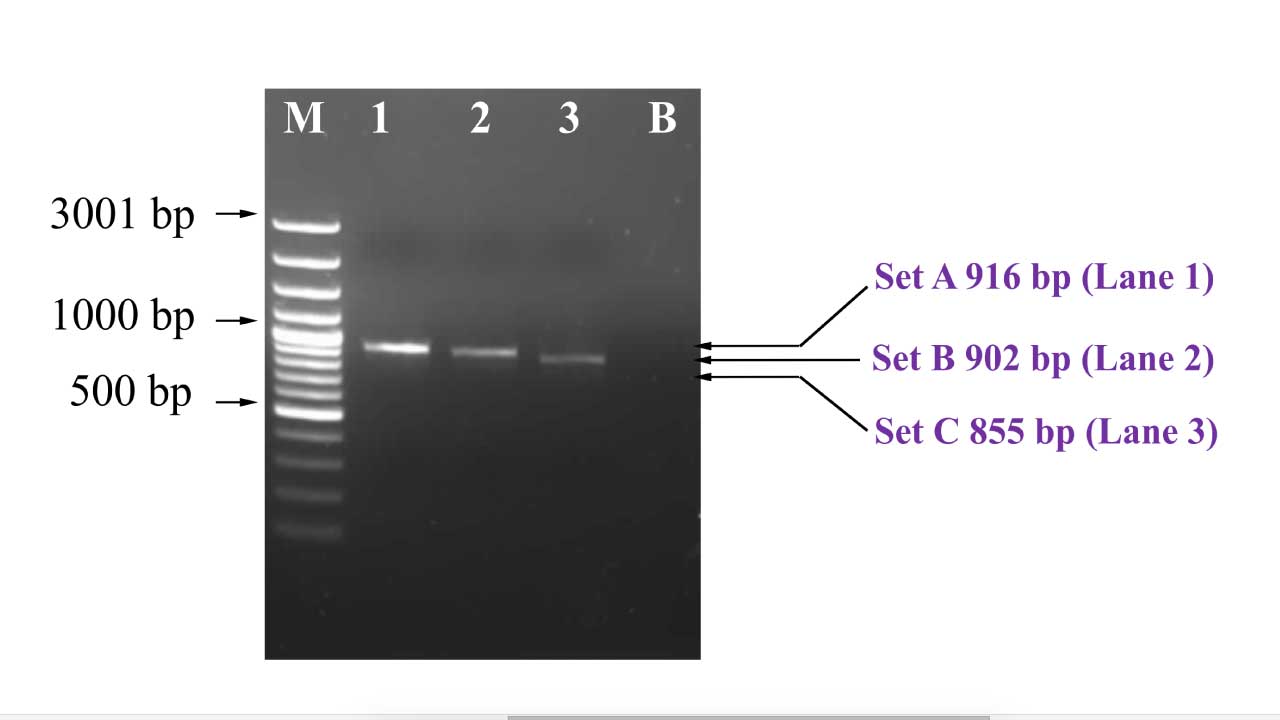
A clean result shows a single bright band at the expected size for each primer set: 916 bp for set A, 902 bp for set B, and 855 bp for set C. The no-template control lane should be completely empty.
Look for these features in a good gel:
- A sharp, well-defined band rather than a smear.
- Brightness comparable across patient samples (uneven loading suggests a pipetting error).
- No band in the no-template control lane.
- No bright cluster of small products at the bottom of the gel, which would indicate primer dimers.
From Gel to Sequence Report
Once products are confirmed, they are purified (using either an enzymatic clean-up like ExoSAP-IT or a column-based kit) and sent for Sanger sequencing. The resulting chromatograms are aligned to the reference HBB sequence (NCBI accession NG_000007.3 or similar) using software such as SnapGene, BioEdit, or Mutation Surveyor.
Variants are then reported using standard HGVS nomenclature and cross-checked against the HbVar and IthaGenes databases to determine clinical significance [7]. A heterozygous mutation appears on the chromatogram as a double peak at the affected base; a homozygous mutation shows a clean single peak that differs from the reference.
Limitations of This Protocol
Direct sequencing of HBB is powerful but not complete. It detects point mutations, small insertions, and small deletions inside the amplified region. It does not detect:
- Large deletions or duplications affecting the gene (these need MLPA or gap-PCR) [4].
- Mutations in regulatory regions outside the amplified segments.
- Low-level mosaicism, where a mutation is present in only a small fraction of cells.
For comprehensive hemoglobinopathy diagnosis, the field is rapidly shifting toward targeted Next-Generation Sequencing (NGS) panels and Third-Generation Long-Read Sequencing (e.g., PacBio, Oxford Nanopore). Long-read sequencing can capture the entire alpha and beta globin clusters in continuous segments, identifying single nucleotide variants, deep intronic mutations, and large structural deletions in a single assay [9]. This effectively replaces the need to run separate Sanger and MLPA workflows in high-throughput centers.
Troubleshooting
Conventional PCR protocol can be a delicate process, and various factors can influence its success. Here are some common issues and potential solutions:
Remember
Troubleshooting PCR protocol often involves a systematic approach. By carefully analyzing the problem and implementing the appropriate solutions, you can significantly improve your PCR success rate.
When to Use This PCR Protocol
This is not a first-line test. The usual diagnostic pathway for suspected beta thalassemia is:
- Full blood count and red cell indices (low MCV, low MCH).
- Hemoglobin analysis by HPLC or capillary electrophoresis (raised HbA2 in carriers).
- ARMS-PCR for common local mutations.
- Direct sequencing of HBB when ARMS-PCR is negative but the clinical and laboratory picture still points to beta thalassemia.
This protocol also has a place in research and in confirming results from newer NGS panels.
Frequently Asked Questions (FAQs)
What is a PCR protocol used for in thalassemia diagnosis?
A PCR protocol amplifies a specific section of DNA so it can be analyzed. In thalassemia diagnosis, PCR copies the HBB gene from a patient's blood sample, producing enough material for direct sequencing. The sequence is then compared against a reference to find mutations that cause reduced or absent beta-globin production.
When should direct sequencing be used instead of ARMS-PCR?
ARMS-PCR is fast and inexpensive but only detects a fixed list of known mutations common to a region. Direct sequencing of HBB is used when ARMS-PCR is negative but the patient still has clinical and laboratory features of thalassemia (low MCV, low MCH, raised HbA2). Sequencing also helps identify rare or novel mutations in patients from genetically diverse populations.
Why is the HBB gene amplified in three overlapping segments?
The HBB gene is roughly 1.6 kb long, including its untranslated regions. Standard PCR works best for fragments under about 1 kb. Splitting the gene into three overlapping amplicons of around 900 bp each (sets A, B, and C) keeps each reaction efficient while still covering the entire gene, so no mutations are missed.
Can this PCR protocol detect alpha thalassemia?
No. This PCR protocol targets the HBB gene on chromosome 11, which is the gene affected in beta thalassemia. Alpha thalassemia involves the HBA1 and HBA2 genes on chromosome 16, and most alpha thalassemia cases are caused by large deletions. These are detected using gap-PCR or MLPA rather than direct sequencing.
Why is a no-template control essential in every PCR run?
The no-template control is a tube containing every reagent except the patient's DNA. If a band appears in this control on the gel, it means contamination has occurred, and the entire run must be discarded. Running this control every time is a basic quality-assurance step in any molecular diagnostics laboratory.
What are the limitations of using Sanger sequencing for beta thalassemia?
Sanger sequencing detects point mutations, small insertions, and small deletions in the amplified region. It does not detect large deletions, rearrangements, or mutations in regions outside the amplified segments. It also has limited sensitivity for low-level mosaicism. For these cases, MLPA, gap-PCR, or next-generation sequencing panels are used alongside or instead of Sanger sequencing.
Glossary of Related Medical Terms
- Allele: One of two or more versions of a gene. A person inherits one allele from each parent.
- Amplicon: The DNA fragment produced by PCR amplification.
- Annealing: The PCR step where primers bind to their matching sequence on the template DNA.
- Beta-globin (HBB): The protein subunit of adult hemoglobin that is reduced or absent in beta thalassemia.
- Chromatogram: The graph produced by Sanger sequencing showing colored peaks for each DNA base.
- Compound heterozygote: A person carrying two different mutations, one on each copy of a gene.
- Denaturation: The PCR step where heat separates the two strands of DNA.
- Exon: A coding region of a gene that contributes to the final protein.
- Extension: The PCR step where DNA polymerase builds a new strand using the primer and template.
- Heterozygous: Carrying two different versions of a gene.
- Homozygous: Carrying two identical versions of a gene.
- Intron: A non-coding region of a gene. Some thalassemia mutations sit inside introns.
- MLPA: Multiplex Ligation-dependent Probe Amplification, a technique for detecting large deletions and duplications.
- PCR protocol: A defined procedure for amplifying a specific DNA segment using primers, polymerase, and cycles of heating and cooling.
- Primer: A short DNA sequence that marks the start point for DNA polymerase during PCR.
- Sanger sequencing: A DNA sequencing method that reads one fragment at a time and produces a chromatogram.
- SNP: Single Nucleotide Polymorphism, a variation at a single DNA base.
- Variant: Any change in DNA sequence compared to a reference.
Disclaimer: This protocol is for educational purposes only. Local laboratory standard operating procedures take precedence. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. Always consult a qualified healthcare professional for clinical decision-making. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Origa R. (2017). β-Thalassemia. Genetics in medicine : official journal of the American College of Medical Genetics, 19(6), 609–619. https://doi.org/10.1038/gim.2016.173
- Cao, A., & Galanello, R. (2010). Beta-thalassemia. Genetics in medicine : official journal of the American College of Medical Genetics, 12(2), 61–76. https://doi.org/10.1097/GIM.0b013e3181cd68ed
- Old J. M. (2003). Screening and genetic diagnosis of haemoglobin disorders. Blood reviews, 17(1), 43–53. https://doi.org/10.1016/s0268-960x(02)00061-9
- Traeger-Synodinos, J., Harteveld, C., Old, J. et al. EMQN Best Practice Guidelines for molecular and haematology methods for carrier identification and prenatal diagnosis of the haemoglobinopathies. Eur J Hum Genet 23, 426–437 (2015). https://doi.org/10.1038/ejhg.2014.131
- Farmakis, D., Porter, J., Taher, A., Domenica Cappellini, M., Angastiniotis, M., & Eleftheriou, A. (2022). 2021 Thalassaemia International Federation Guidelines for the Management of Transfusion-dependent Thalassemia. HemaSphere, 6(8), e732. https://doi.org/10.1097/HS9.0000000000000732
- Shang, X., & Xu, X. (2017). Update in the genetics of thalassemia: What clinicians need to know. Best practice & research. Clinical obstetrics & gynaecology, 39, 3–15. https://doi.org/10.1016/j.bpobgyn.2016.10.012
- Giardine, B., Borg, J., Viennas, E., Pavlidis, C., Moradkhani, K., Joly, P., Bartsakoulia, M., Riemer, C., Miller, W., Tzimas, G., Wajcman, H., Hardison, R. C., & Patrinos, G. P. (2014). Updates of the HbVar database of human hemoglobin variants and thalassemia mutations. Nucleic acids research, 42(Database issue), D1063–D1069. https://doi.org/10.1093/nar/gkt911
- McInerney, P., Adams, P., & Hadi, M. Z. (2014). Error Rate Comparison during Polymerase Chain Reaction by DNA Polymerase. Molecular biology international, 2014, 287430. https://doi.org/10.1155/2014/287430
- Xu, L., Mao, A., Liu, H., Gui, B., Choy, K. W., Huang, H., Yu, Q., Zhang, X., Chen, M., Lin, N., Chen, L., Han, J., Wang, Y., Zhang, M., Li, X., He, D., Lin, Y., Zhang, J., Cram, D. S., & Cao, H. (2020). Long-Molecule Sequencing: A New Approach for Identification of Clinically Significant DNA Variants in α-Thalassemia and β-Thalassemia Carriers. The Journal of molecular diagnostics : JMD, 22(8), 1087–1095. https://doi.org/10.1016/j.jmoldx.2020.05.004
- Lim, W. F., Muniandi, L., George, E., Sathar, J., Teh, L. K., Gan, G. G., & Lai, M. I. (2012). α-Haemoglobin stabilising protein expression is influenced by mean cell haemoglobin and HbF levels in HbE/β-thalassaemia individuals. Blood cells, molecules & diseases, 48(1), 17–21. https://doi.org/10.1016/j.bcmd.2011.10.002