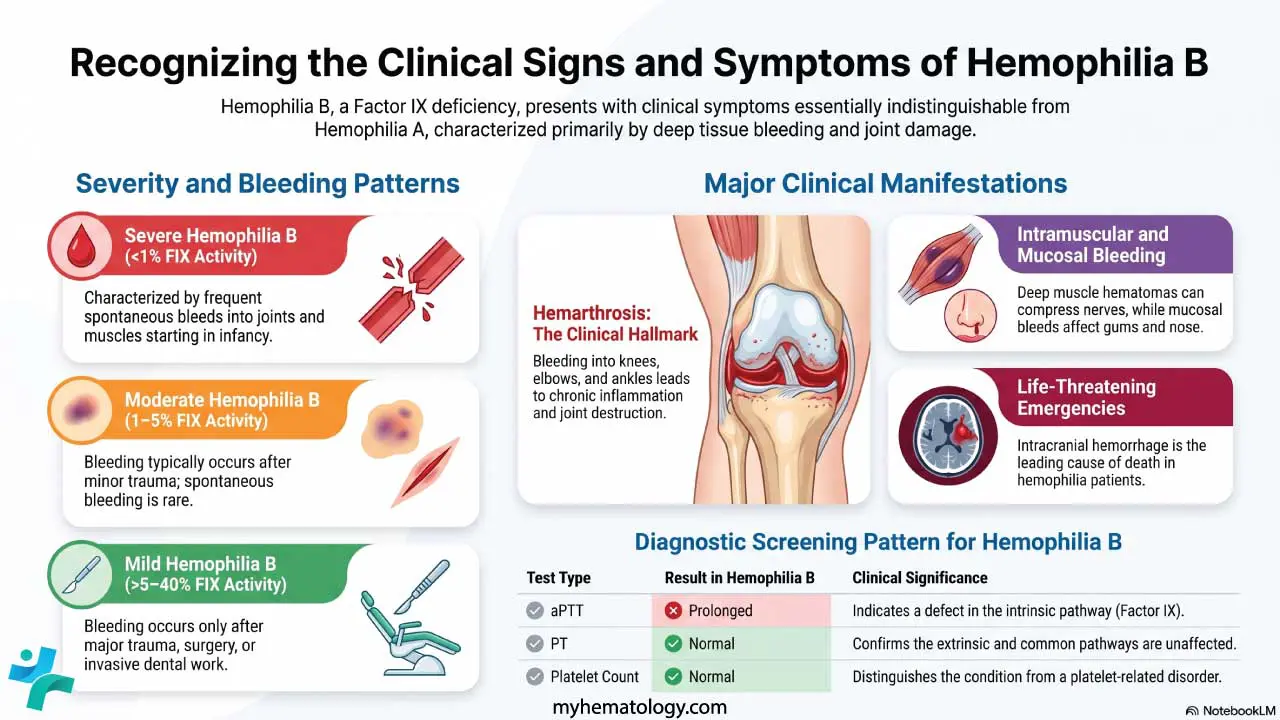
Key Takeaways
Primary immune thrombocytopenia (ITP) is an acquired autoimmune disorder in which the immune system both destroys platelets and slows their production, dropping the platelet count below 100 × 10⁹/L with no other cause found [1,2].
- Symptoms ▾: Common signs are petechiae (pinpoint red spots), easy bruising, nosebleeds, gum bleeding, and heavy periods. Intracranial hemorrhage is rare but is the leading cause of ITP-related death [1].
- Diagnosis ▾: Diagnosis is by exclusion. A complete blood count shows isolated low platelets; further tests rule out infections, lupus, thyroid disease, and other mimics. Bone marrow biopsy is not routinely required [1,2].
- Treatment ▾: Most guidelines start treatment if platelets fall below 30 × 10⁹/L or if clinically significant bleeding occurs [1]. First-line therapy is a short course of corticosteroids, with IVIg or anti-D used when a faster effect is needed. Second-line options include TPO receptor agonists, rituximab, and newer agents such as fostamatinib, rilzabrutinib, and efgartigimod [1,3,4,5].
*Click ▾ for more information
What is Primary Immune Thrombocytopenia?
Primary immune thrombocytopenia (ITP) is an autoimmune disorder in which the body's own immune system targets healthy platelets, the small cell fragments that plug damaged blood vessels and stop bleeding. The platelet count drops below 100 × 10⁹/L, while the rest of the blood cell counts stay normal [1].
You may still see this condition called idiopathic thrombocytopenic purpura or chronic idiopathic thrombocytopenic purpura in older textbooks. Hematology guidelines now prefer primary immune thrombocytopenia because the immune mechanism is well established, and the word "primary" makes clear that no other underlying disease is driving the platelet drop [1,2].
Primary vs. Secondary ITP
Primary ITP stands alone — low platelets with no other explanation. Secondary ITP looks similar but is driven by something else: HIV, hepatitis C, Helicobacter pylori, systemic lupus erythematosus (SLE), or certain drugs. Distinguishing the two is the central diagnostic task, because treating the underlying cause may resolve the thrombocytopenia [1,2].
How ITP Damages Platelets
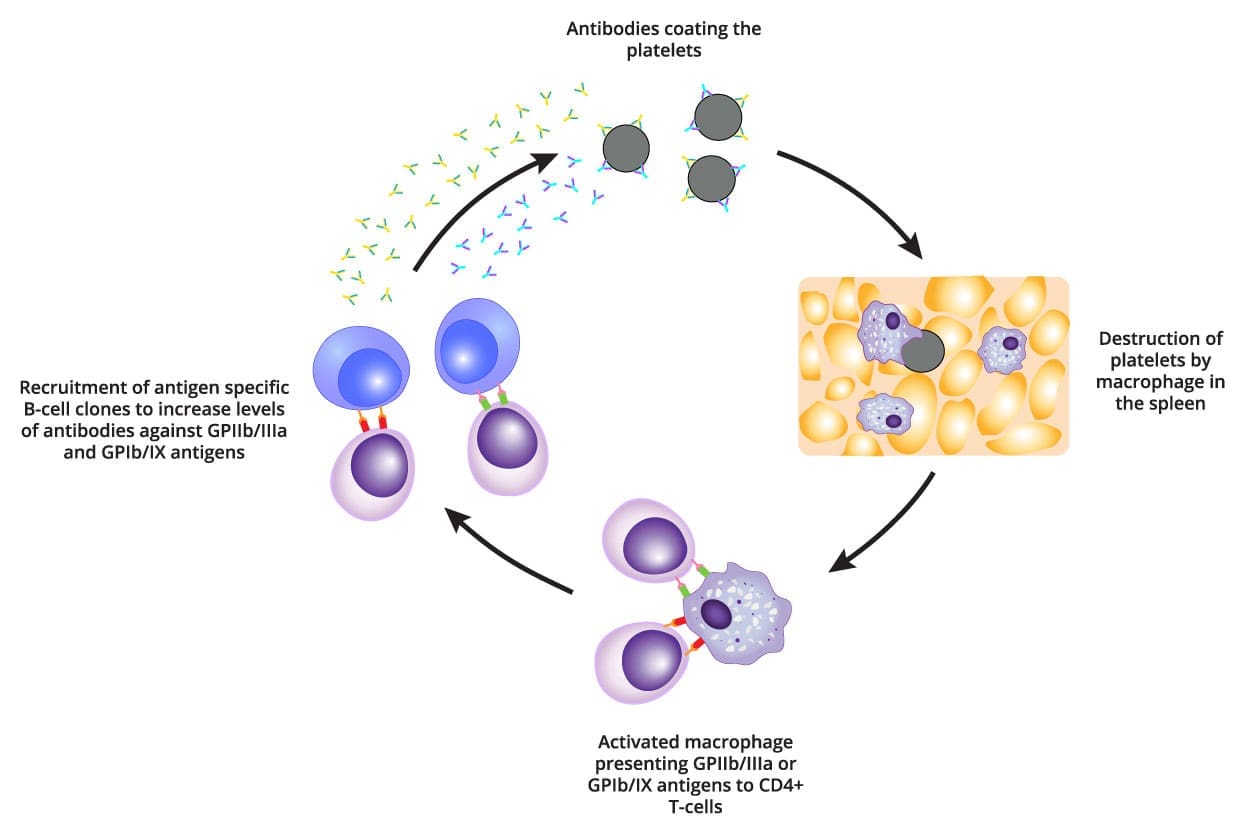
For decades, ITP was taught as a destruction problem only. The modern picture is a dual defect: immune destruction of mature platelets and impaired production of new ones [6].
Destruction in the Spleen
B cells produce IgG autoantibodies — antibodies aimed mistakenly at the body's own tissue. In ITP these autoantibodies bind to surface proteins on platelets, most often glycoprotein IIb/IIIa and glycoprotein Ib/IX. Once a platelet is coated (a process called opsonization), it is recognized by macrophages, the cleanup cells of the spleen and liver, and removed by phagocytosis [6].
This rogue immune behavior is supported by an underlying T-cell imbalance. Pro-inflammatory helper T cells (Th1 and Th17) (the ones that drive aggressive immune responses) are increased. Regulatory T cells (Tregs) (the brakes that normally hold autoimmunity in check) are reduced [6].
Impaired Production in the Bone Marrow
The same autoantibodies also attack megakaryocytes, the giant marrow cells that bud off platelets. They suppress megakaryocyte maturation and push these cells toward apoptosis (programmed cell death). Even when megakaryocyte numbers look adequate or increased on biopsy, their output of platelets falls short of demand [6].
Clinical Features
Symptoms of primary immune thrombocytopenia track closely with the platelet count, but with wide individual variation. Some patients are diagnosed incidentally on a routine blood test and feel completely well [1].
Common Bleeding Manifestations
Visible bleeding usually appears below 50 × 10⁹/L and becomes worrying below 20 × 10⁹/L.
- Petechiae: Pinpoint reddish-purple spots, 1–2 mm, most often on the lower legs. They are the classic sign.
- Purpura: Larger bruise-like patches from bleeding under the skin, often appearing without trauma.
- Mucosal Bleeding: Nosebleeds (epistaxis), gum bleeding when brushing, and occasionally gastrointestinal bleeding.
- Menorrhagia: Heavy or prolonged periods, which can lead to iron deficiency anemia.
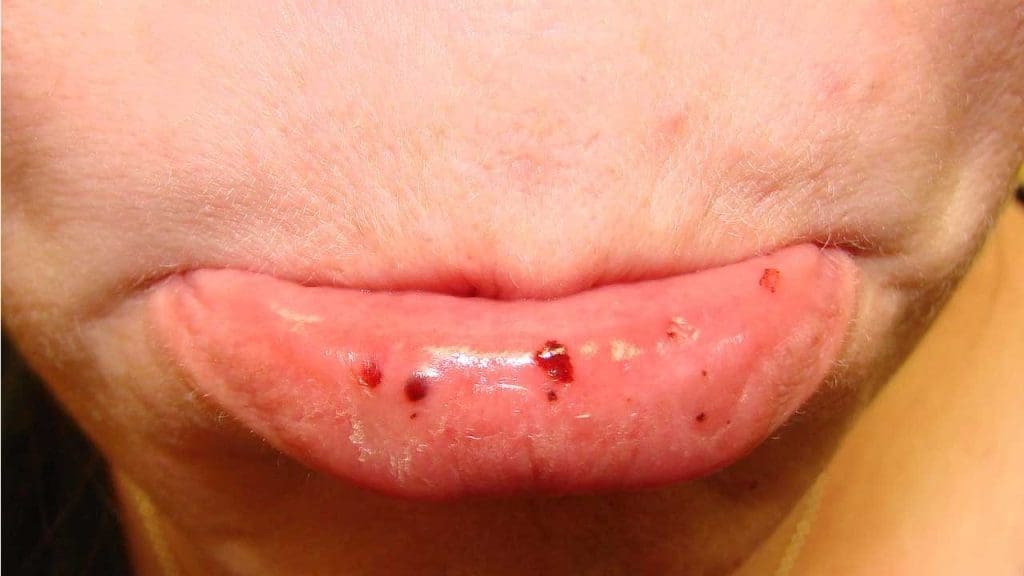
Severe and Life-Threatening Bleeding
Severe internal bleeding becomes a real risk below 10 × 10⁹/L.
- Gastrointestinal bleeding: Vomiting blood (hematemesis) or black, tarry stool (melena).
- Intracranial hemorrhage (ICH): Rare (<1% of adults) but the leading cause of ITP-related death [1]. Sudden severe headache, vision or speech changes, or new neurological signs need immediate emergency assessment.
What Caregivers Can Watch For at Home
For families supporting a patient with primary immune thrombocytopenia, the signs that warrant urgent care are: bleeding that won't stop after 10 minutes of pressure, blood in vomit or stool, a new or "thunderclap" headache, confusion, or new weakness on one side of the body. Day-to-day, the things that quietly raise risk are over-the-counter painkillers (aspirin, ibuprofen, naproxen) and contact sports.
The Symptoms No One Sees
Up to two-thirds of patients with chronic ITP report significant fatigue and mood disturbance, even when their platelet counts are reasonable [8]. The unpredictability of bleeding episodes, the need for frequent blood tests, and the side effects of treatment all weigh on quality of life. Validated tools such as ITP-PAQ help clinicians measure this burden and adjust care.
Clinical Assessment and Grading the Bleeding
Standardized scoring systems like the WHO Bleeding Score and ITP-specific tools let clinicians grade severity from 0 (none) to 4 (life-threatening). The threshold for treatment is generally a platelet count below 30 × 10⁹/L or clinically significant bleeding regardless of count [1].
How IPT is Diagnosed
There is no single test that confirms ITP. The diagnosis is reached by finding isolated thrombocytopenia and excluding everything else [1,2].
Core Tests Everyone Gets
- Complete blood count (CBC): Should show low platelets only. If hemoglobin or white cell counts are also abnormal, suspicion shifts toward marrow disease or another systemic problem.
- Peripheral blood smear: Confirms true thrombocytopenia. It often shows large young platelets (megathrombocytes) released early from a stressed marrow. The smear also rules out pseudothrombocytopenia, in which platelets clump in EDTA tubes and falsely appear low.
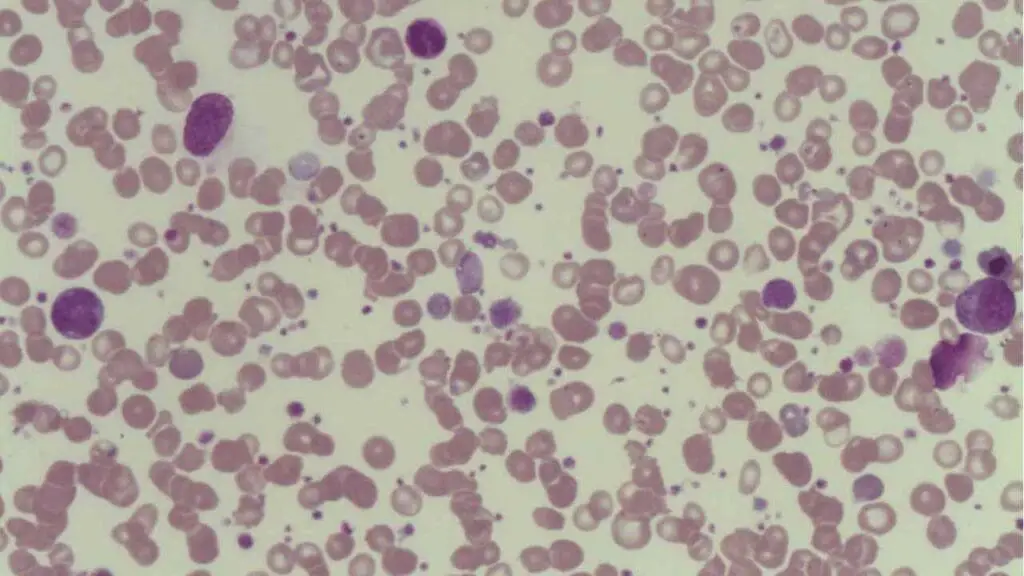
Tests to Rule Out Secondary ITP
Each can cause low platelets directly or via immune cross-reaction. H. pylori eradication can resolve ITP in some patients.
Screens for lupus, autoimmune thyroid disease, and related disorders.
A positive DAT suggests that autoantibodies are also targeting red blood cells, pointing toward Evans Syndrome (ITP + Autoimmune Hemolytic Anemia).
Specialized and Confirmatory Tests
These tests are typically reserved for specific clinical scenarios or research, and are not required for the initial diagnosis of Primary Immune Thrombocytopenia in a classic presentation.
- Antiplatelet antibody assays are highly specific but only moderately sensitive. A negative result does not rule out ITP, so they are not part of routine workup [1,2].
- Bone marrow biopsy is not needed for typical adult or pediatric ITP. It is considered when the picture is atypical: age over 60 (to exclude myelodysplastic syndrome), other cytopenias, failure of first-line treatment, or sometimes before splenectomy.
Common Diagnostic Pitfalls
The three traps we should remember are pseudothrombocytopenia (clumping artifact), drug-induced thrombocytopenia (heparin, quinine, sulfa drugs, vancomycin), and TTP (thrombotic thrombocytopenic purpura), which is a hematologic emergency. A blood smear showing schistocytes points to TTP, not ITP.
Differential Diagnosis
Platelet Disorders
|
Secondary ITP SLE · HIV · HCV · H. pylori · CLL |
Immune destruction
Immune destruction driven by underlying disease
|
Positive serology or other clinical features of the underlying condition |
| Myelodysplastic Syndromes (MDS) |
Ineffective hematopoiesis
Ineffective hematopoiesis
|
Dysplastic cells on blood smear; abnormal marrow on biopsy |
| B12 / Folate Deficiency |
Ineffective hematopoiesis
Impaired DNA synthesis
|
Macrocytic anemia, low B12 or folate levels |
| Aplastic Anemia |
Ineffective hematopoiesis
Destruction of hematopoietic stem cells
|
Pancytopenia; hypocellular marrow on biopsy |
| Thrombotic Thrombocytopenic Purpura (TTP) |
Consumptive
Microvascular platelet consumption
|
Schistocytes on smear; neurological or kidney signs |
| Disseminated Intravascular Coagulation (DIC) |
Consumptive
Widespread consumption of platelets and clotting factors
|
High D-dimer; prolonged PT / aPTT |
|
Hypersplenism e.g., in Cirrhosis |
Sequestration
Splenic sequestration of platelets
|
Splenomegaly on imaging; abnormal liver function tests |
| Pseudothrombocytopenia |
Artifact
EDTA-induced platelet clumping in vitro
|
Platelet clumps visible on smear; normal count in citrate tube |
|
Inherited Thrombocytopenias e.g., Bernard-Soulier Syndrome |
Genetic defect
Genetic platelet defect
|
Lifelong history, giant platelets on smear, positive family history |
| Drug-induced Thrombocytopenia |
Drug-related
Drug-dependent antibodies or direct marrow toxicity
|
Recent exposure to offending drug (heparin, quinine, vancomycin, sulfa) |
| COVID-19 / Vaccine-associated ITP |
Post-infectious / immune
Post-infectious or post-vaccination immune trigger
|
Recent COVID-19 illness or vaccination history |
Treatment & Management
The goal of primary immune thrombocytopenia treatment is not to push platelets back to normal. It is to prevent clinically meaningful bleeding while minimizing drug side effects [1].
Treatment is typically started when the platelet count is below 30 × 10⁹/L, or whenever significant bleeding occurs, regardless of count [1,2].
| Drug | Mechanism of Action | Clinical Notes |
|---|---|---|
| Corticosteroids Prednisone or pulsed dexamethasone | Suppress autoantibody production and block macrophage Fc receptors | Modern guidelines favor short courses (≤6 weeks) to avoid weight gain, diabetes, mood changes, and bone loss [1,2] |
| Intravenous Immunoglobulin IVIg | Saturates macrophage Fc receptors so they ignore antibody-coated platelets |
Acts within days but effect lasts only days to weeks; used before surgery, delivery, or in major bleeds
Rapid but transient response |
| Anti-D Immunoglobulin Rho(D) Immune Globulin | Causes mild RBC coating that distracts macrophages from platelets |
Only for Rh-positive, non-splenectomized patients; carries an FDA black-box warning for severe hemolysis — use is now selective
FDA Black-Box Warning: Severe Hemolysis |
Second-Line Therapy (Chronic Management)
If platelets stay low after about three months, or if relapse follows a steroid taper, second-line therapy enters the picture. Current clinical guidelines strongly prioritize thrombopoietin receptor agonists (TPO-RAs) as the preferred first choice for second-line treatment. Rituximab is now generally deferred due to its lower long-term remission rates and immunosuppressive risks [1,7,9].
Thrombopoietin receptor agonists (TPO-RAs): Mimic the natural hormone thrombopoietin and tell the bone marrow to make more platelets [1].
- Romiplostim: weekly subcutaneous injection.
- Eltrombopag: oral; must be taken away from calcium, iron, and antacids.
- Avatrombopag: oral; no food restrictions. TPO-RAs work in about 60–80% of chronic ITP patients but raise thrombosis risk and require periodic monitoring.
Rituximab: A monoclonal antibody against CD20 that depletes B cells. While about 60% of patients respond initially, durable remissions are seen in fewer than 30%. Because it suppresses the immune system, modern practice typically reserves it for later lines of therapy [1,9].
Fostamatinib (Tavalisse): An oral SYK (spleen tyrosine kinase) inhibitor approved by the FDA in 2018 for chronic ITP after insufficient response to a prior therapy. It blocks the macrophage signaling that drives platelet destruction [3].
Rilzabrutinib (Wayrilz): An oral BTK inhibitor. Following the highly successful phase 3 LUNA3 trial, it is now an approved, targeted option for adult patients with persistent or chronic ITP who have had an insufficient response to prior therapies [4,10].
Efgartigimod: An IV FcRn antagonist that lowers circulating IgG, including the autoantibodies that drive ITP. The phase 3 ADVANCE-IV trial showed a sustained platelet response in heavily pretreated chronic ITP; however, it is important to note that while approved for other autoimmune conditions, it remains investigational for ITP pending final regulatory decisions [5].
Splenectomy: Removes the main site of platelet destruction. Although it offers a high long-term remission rate, modern practice increasingly avoids it altogether. With the advent of highly effective TPO-RAs and newer targeted inhibitors, surgery is now largely viewed as a last-resort option [1,9].
Treating the Underlying Cause
If H. pylori infection is found, eradication therapy can normalize platelets in some patients without immunosuppression [1].
Emergency and Refractory ITP
For life-threatening bleeding, hematologists combine high-dose IVIg, IV corticosteroids, and platelet transfusions. Transfused platelets are destroyed quickly too, but they can buy critical time.Refractory ITP is a disease that persists despite splenectomy and at least one second-line drug and that usually means combining therapies, rotating among the newer agents, or enrolling in clinical trials [7].
Special Populations & Safety Considerations
Pregnancy
Primary immune thrombocytopenia is one of the most common causes of thrombocytopenia in pregnancy, after gestational thrombocytopenia. Treatment decisions hinge on maternal bleeding risk, not the fetal platelet count [1,2].
- First-line: corticosteroids (often prednisone).
- Near delivery: IVIg if a rapid bump is needed.
- Avoid: TPO-RAs and rituximab, particularly in the first trimester, because of limited fetal safety data.
- Mode of delivery is determined by obstetric indications. Routine fetal platelet monitoring is generally not recommended because severe neonatal thrombocytopenia is uncommon.
Children
Pediatric primary immune thrombocytopenia (typically ages 2–6) usually behaves very differently from the adult disease.
- Around 80% of cases resolve spontaneously within months [1].
- Observation alone is appropriate for most children with platelets above 20 × 10⁹/L and no significant bleeding.
- Treatment (IVIg, anti-D, or short corticosteroid course) is reserved for platelets below 10 × 10⁹/L or significant mucosal bleeding.
- Intracranial hemorrhage risk in pediatric ITP is very low (around 0.5%) [2].
- About 20% develop chronic ITP and follow adult-style management.
Older Adults: The Bleeding-vs-Clotting Balance
Patients over 60 face a double-edged challenge. They bleed more readily at low platelet counts, and they are more likely to have cardiovascular disease that itself raises clotting risk. TPO-RAs add to thrombosis risk. Treatment in this group is genuinely individualized [1,2].
Post-Splenectomy Safety
The spleen filters bloodborne bacteria, especially encapsulated organisms. Without it, patients face a small but real risk of overwhelming post-splenectomy infection (OPSI).
- Vaccinate before surgery (ideally several weeks ahead): pneumococcal (PCV13/PCV15 plus PPSV23), meningococcal ACWY and B, and Haemophilus influenzae type B.
- Antibiotic prophylaxis (penicillin or amoxicillin) is often prescribed for at least the first few years and sometimes lifelong.
- Educate patients to seek urgent care for any unexplained fever.
COVID-19 and Vaccine-Associated ITP
Both SARS-CoV-2 infection and several COVID-19 vaccines have been linked to new or relapsed ITP [7]. The mechanism is thought to be immune dysregulation triggered by the infection or the immune response to vaccination. Onset is usually within weeks. Most cases respond to standard ITP therapy.
Complications and Prognosis
What Can Go Wrong
- Intracranial hemorrhage: Rare overall but the leading cause of ITP-related death; risk rises sharply below 10 × 10⁹/L [1].
- Severe mucosal bleeding: Heavy nasal, gum, or GI bleeding can require emergency platelet transfusion.
- Iron deficiency anemia: Especially in women with menorrhagia.
- OPSI: Lifelong risk after splenectomy.
- Treatment side effects: Corticosteroids cause weight gain, hypertension, diabetes, and bone loss with long use; TPO-RAs raise thrombosis risk and can cause mild marrow reticulin changes.
For most adults, primary immune thrombocytopenia is a chronic condition that is managed, not cured. With modern therapy, the great majority of patients live full lives at platelet counts that keep them safe from serious bleeding [1,7].
Frequently Asked Questions (FAQs)
What is primary immune thrombocytopenia?
Primary immune thrombocytopenia (ITP) is an autoimmune disorder in which the immune system destroys platelets and slows their production in the bone marrow, dropping the platelet count below 100 × 10⁹/L without any other identifiable cause. Older names for the same condition include idiopathic thrombocytopenic purpura and chronic ITP.
How is primary immune thrombocytopenia diagnosed?
Diagnosis is by exclusion. A complete blood count shows isolated low platelets, and the blood smear confirms genuine thrombocytopenia. Doctors then test for secondary causes such as HIV, hepatitis C, H. pylori, lupus, and thyroid disease. A bone marrow biopsy is not routinely needed but may be done if the patient is older, has other low blood counts, or fails first-line treatment.
What platelet count is dangerous in ITP?
Bleeding risk rises noticeably below 30 × 10⁹/L and becomes serious below 10 × 10⁹/L, where the rare but life-threatening complication of intracranial hemorrhage can occur. Most guidelines recommend starting treatment when the platelet count falls below 30 × 10⁹/L or when significant bleeding is present, regardless of count.
What are the newer treatments for chronic ITP?
Beyond corticosteroids, IVIg, and TPO receptor agonists (romiplostim, eltrombopag, avatrombopag), newer options for chronic or refractory ITP include fostamatinib (an oral SYK inhibitor), rilzabrutinib (an oral BTK inhibitor), and efgartigimod (an IV FcRn antagonist that lowers harmful autoantibodies). Rituximab and, less often, splenectomy remain second-line choices.
Can children outgrow ITP?
Yes, frequently. About 80% of pediatric ITP cases resolve spontaneously within 6–12 months, which is why most children with mild disease are simply observed. About 20% develop chronic ITP and need long-term management similar to adults.
Is ITP inherited or contagious?
ITP is not contagious and is not classically inherited. Most cases are sporadic. There is a small genetic predisposition — close relatives of someone with ITP have a slightly higher risk of autoimmune disease in general, but ITP itself is not passed down in a predictable pattern.
Which over-the-counter medications should I avoid with low platelets?
Avoid drugs that further impair platelet function: aspirin and NSAIDs such as ibuprofen (Advil, Motrin) and naproxen (Aleve). Acetaminophen (paracetamol) is generally a safer alternative for pain or fever, but always check with the treating hematologist before starting any new medication.
Glossary of Related Medical Terms
- Apoptosis: Programmed cell death — a controlled "self-destruct" sequence that cells use to die without harming surrounding tissue.
- Autoantibody: An antibody the immune system makes by mistake against the body's own cells. In ITP, autoantibodies tag platelets for destruction.
- BTK (Bruton tyrosine kinase) inhibitor: A drug class (e.g., rilzabrutinib) that blocks an enzyme used by B cells, dampening autoantibody production.
- Cytopenia: A low count of any blood cell type. ITP causes one cytopenia — low platelets — by definition.
- Epistaxis: A nosebleed.
- FcRn antagonist: A drug (e.g., efgartigimod) that lowers circulating IgG antibodies, including the harmful autoantibodies in ITP.
- Glycoprotein IIb/IIIa: A protein on the platelet surface; the most common autoantibody target in ITP.
- Intracranial hemorrhage (ICH): Bleeding inside the skull. The most feared ITP complication.
- Macrophage: A "clean-up" white blood cell, mainly in the spleen and liver, that engulfs antibody-coated platelets in ITP.
- Megakaryocyte: The large bone marrow cell that produces platelets.
- Menorrhagia: Heavy or prolonged menstrual bleeding.
- Opsonization: The "tagging" of a cell with antibody so immune cells will recognize and remove it.
- Petechiae: Tiny pinpoint red-purple spots from bleeding under the skin; a hallmark of low platelets.
- Primary immune thrombocytopenia (ITP): An autoimmune disorder in which the immune system destroys and underproduces platelets, with no other identifiable cause.
- Purpura: Larger bruise-like skin patches caused by bleeding under the skin.
- Splenectomy: Surgical removal of the spleen.
- SYK inhibitor: A drug (e.g., fostamatinib) that blocks spleen tyrosine kinase, the enzyme macrophages use to destroy antibody-coated platelets.
- Thrombocytopenia: A platelet count below the normal range (commonly defined as <150 × 10⁹/L; <100 × 10⁹/L for ITP).
- TPO-RA (thrombopoietin receptor agonist): A drug class (romiplostim, eltrombopag, avatrombopag) that mimics natural thrombopoietin and pushes the bone marrow to make more platelets.
Disclaimer: This article is intended for educational and informational purposes only. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Neunert, C., Terrell, D. R., Arnold, D. M., Buchanan, G., Cines, D. B., Cooper, N., Cuker, A., Despotovic, J. M., George, J. N., Grace, R. F., Kühne, T., Kuter, D. J., Lim, W., McCrae, K. R., Pruitt, B., Shimanek, H., & Vesely, S. K. (2019). American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood advances, 3(23), 3829–3866. https://doi.org/10.1182/bloodadvances.2019000966
- Provan, D., Arnold, D. M., Bussel, J. B., Chong, B. H., Cooper, N., Gernsheimer, T., Ghanima, W., Godeau, B., González-López, T. J., Grainger, J., Hou, M., Kruse, C., McDonald, V., Michel, M., Newland, A. C., Pavord, S., Rodeghiero, F., Scully, M., Tomiyama, Y., Wong, R. S., … Kuter, D. J. (2019). Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood advances, 3(22), 3780–3817. https://doi.org/10.1182/bloodadvances.2019000812
- Bussel, J., Arnold, D. M., Grossbard, E., Mayer, J., Treliński, J., Homenda, W., Hellmann, A., Windyga, J., Sivcheva, L., Khalafallah, A. A., Zaja, F., Cooper, N., Markovtsov, V., Zayed, H., & Duliege, A. M. (2018). Fostamatinib for the treatment of adult persistent and chronic immune thrombocytopenia: Results of two phase 3, randomized, placebo-controlled trials. American journal of hematology, 93(7), 921–930. https://doi.org/10.1002/ajh.25125
- Kuter, D. J., Ghanima, W., Cooper, N., Liebman, H. A., Zhang, L., Hu, Y., Miyakawa, Y., Homenda, W., Galindo, L. E. M., Basquiera, A. L., Tan, C. W., Saydam, G., Hütter-Krönke, M. L., Chai-Adisaksopha, C., Gómez-Almaguer, D., Tran, H., Shin, H. J., Dantas da Cunha Junior, A., Lazar, Z., Izquierdo, C. P., … Daak, A. (2025). Safety and efficacy of rilzabrutinib vs placebo in adults with immune thrombocytopenia: the phase 3 LUNA3 study. Blood, 145(24), 2914–2926. https://doi.org/10.1182/blood.2024027336
- Broome, C., Miyakawa, Y., Carpenedo, M., Al-Samkari, H., Ayguasanosa, J., Phillips, J., & Rodeghiero, F. (2026). Efgartigimod as a treatment for adults with primary immune thrombocytopenia: a plain language summary of the ADVANCE IV study. Therapeutic advances in hematology, 17, 20406207261445668. https://doi.org/10.1177/20406207261445668
- Gernsheimer T. (2009). Chronic idiopathic thrombocytopenic purpura: mechanisms of pathogenesis. The oncologist, 14(1), 12–21. https://doi.org/10.1634/theoncologist.2008-0132
- Gallo, P. M., & Lambert, M. P. (2024). Immune thrombocytopenia guidelines get an annual checkup. Blood advances, 8(13), 3576–3577. https://doi.org/10.1182/bloodadvances.2024013317
- Cooper, N., Kruse, A., Kruse, C., Watson, S., Morgan, M., Provan, D., Ghanima, W., Arnold, D. M., Tomiyama, Y., Santoro, C., Michel, M., Laborde, S., Lovrencic, B., Hou, M., Bailey, T., Taylor-Stokes, G., Haenig, J., & Bussel, J. B. (2021). Immune thrombocytopenia (ITP) World Impact Survey (iWISh): Patient and physician perceptions of diagnosis, signs and symptoms, and treatment. American journal of hematology, 96(2), 188–198. https://doi.org/10.1002/ajh.26045
- Ghanima, W., Cuker, A., & Michel, M. (2024). Insights on treatment of adult ITP: algorithm for management and role of multimodal therapy. Hematology. American Society of Hematology. Education Program, 2024(1), 678–684. https://doi.org/10.1182/hematology.2024000594
- Kuter, D. J., Efraim, M., Mayer, J., Trněný, M., McDonald, V., Bird, R., Regenbogen, T., Garg, M., Kaplan, Z., Tzvetkov, N., Choi, P. Y., Jansen, A. J. G., Kostal, M., Baker, R., Gumulec, J., Lee, E. J., Cunningham, I., Goncalves, I., Warner, M., Boccia, R., … Cooper, N. (2022). Rilzabrutinib, an Oral BTK Inhibitor, in Immune Thrombocytopenia. The New England journal of medicine, 386(15), 1421–1431. https://doi.org/10.1056/NEJMoa2110297