Key Takeaways
Hemophagocytic Lymphohistiocytosis or HLH is a life-threatening "cytokine storm" where a broken "off-switch" in the immune system leads to unchecked, hyperactive T-cells and macrophages that attack the patient's own organs and blood cells.
- Pathophysiology ▾: A failure in the perforin/granzyme cytotoxic pathway means NK and T-cells cannot kill their targets. The persistent antigen triggers massive Interferon-gamma release, turning macrophages into "angry," pathological histiocytes.
- Two Main Classifications ▾:
- Primary (Familial): Genetic mutations in the cytotoxic pathway (e.g., PRF1); typically presents in infancy.
- Secondary (Acquired): Triggered by severe infections (classic: Epstein-Barr Virus), malignancies (T/NK-cell lymphomas), or rheumatologic diseases (Macrophage Activation Syndrome).
- Classic Presentation ▾: Unremitting high fever, massive hepatosplenomegaly, severe cytopenias, abnormal liver function, and neurological decline mimicking severe, culture-negative sepsis.
- Key Investigations ▾:
- Ferritin: Massively elevated (often > 10,000 ng/mL) due to extreme macrophage activation.
- Fibrinogen: Dangerously low (≤ 1.5 g/L) due to hepatic consumption and DIC.
- Triglycerides: Elevated ≥ 3.0 mmol/L) as cytokines inhibit lipoprotein lipase.
- sCD25 (sIL-2R): Highly elevated, reflecting massive, uncontrolled T-cell activation.
- Bone Marrow: Shows the hallmark morphological sign of hemophagocytosis (macrophages eating RBCs/WBCs/platelets).
- Diagnostic Tools ▾: The HLH-2004 criteria (requires 5 of 8 specific clinical/lab markers) or the HScore (a weighted point system calculating the probability of secondary HLH, which uniquely includes AST levels).
- Treatment & Management ▾: A medical emergency requiring the HLH-94 protocol (Etoposide to kill hyperactive T-cells + Dexamethasone to suppress macrophages and cross the blood-brain barrier), alongside trigger-specific therapies (like Rituximab for EBV). Allogeneic Hematopoietic Stem Cell Transplant (HSCT) is the only curative therapy for primary cases or refractory secondary cases.
*Click ▾ for more information
Introduction
Hemophagocytic Lymphohistiocytosis (HLH) is one of the most severe, rapidly progressive, and life-threatening hyperinflammatory syndromes encountered in clinical medicine. Rather than a single specific disease, Hemophagocytic Lymphohistiocytosis (HLH) is best understood as a devastating clinical syndrome representing the extreme end of the immune activation spectrum.
In a healthy immune system, an infection or abnormal cell triggers a precise, regulated response. Once the threat is neutralized, the immune system naturally downregulates to prevent damage to the host's own tissues. In Hemophagocytic Lymphohistiocytosis (HLH), this critical "off switch" is broken.
This uncontrolled hyperinflammation leads to a massive, systemic release of inflammatory cytokines. Often referred to as a "cytokine storm", it rapidly causes progressive multi-organ failure and, if not aggressively treated, death. Recognizing Hemophagocytic Lymphohistiocytosis (HLH) early is paramount, as the initial presentation frequently mimics severe sepsis or shock, requiring a high index of clinical suspicion to intervene in time.
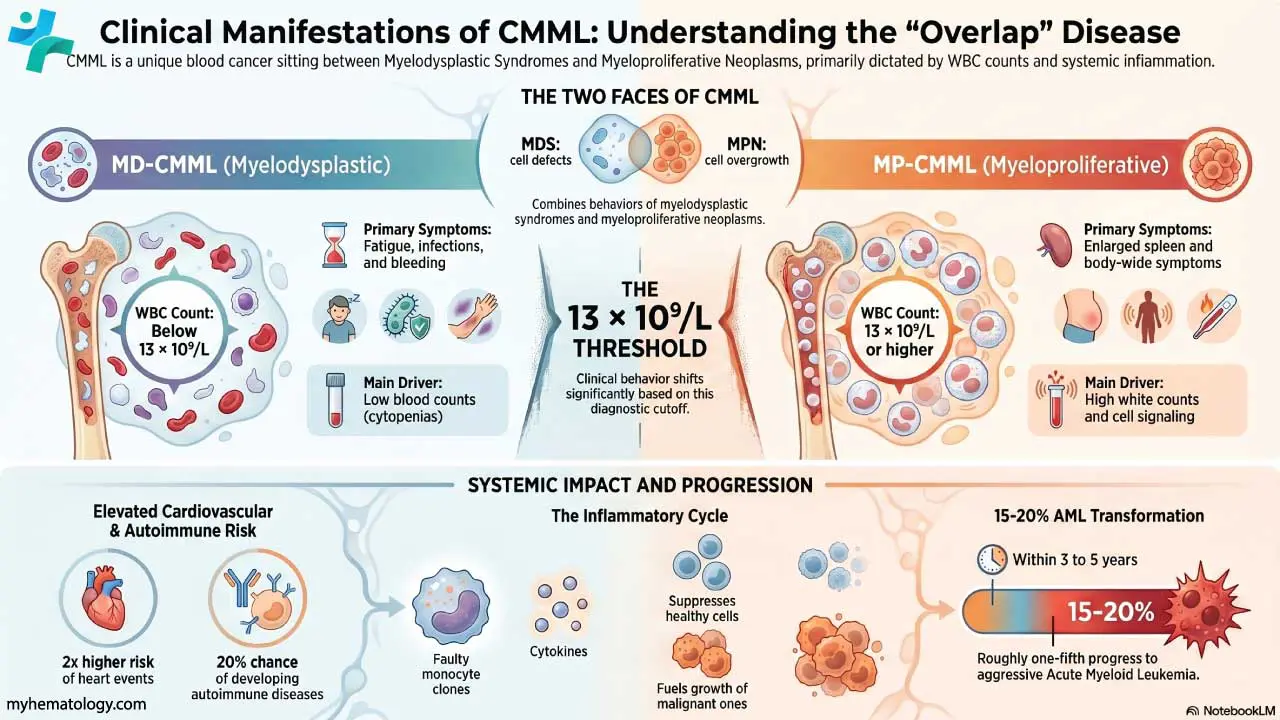
Defining Hemophagocytic Lymphohistiocytosis (HLH)
Clinically and pathophysiologically, Hemophagocytic Lymphohistiocytosis (HLH) is defined by the uncontrolled proliferation and activation of morphologically benign macrophages (histiocytes) and T lymphocytes (specifically CD8+ cytotoxic T-cells).
The core functional defect lies in the impairment of Natural Killer (NK) cells and cytotoxic T-cells. Normally, these cells are responsible for clearing infected cells and terminating the immune response via perforin-mediated cytotoxicity. When this mechanism fails, the immune system cannot clear the initial antigenic trigger. This results in sustained, amplified interaction between T-cells and macrophages, leading to:
- Massive Cytokine Release: Hypersecretion of Interferon-gamma (IFN-γ), Tumor Necrosis Factor-alpha (TNF-α), Interleukin-6 (IL-6), and others, which drive the systemic symptoms like unremitting fever and organ damage.
- Hemophagocytosis: The defining morphological hallmark of the syndrome. The hyperactivated macrophages begin to inappropriately engulf (phagocytose) the patient's own red blood cells, white blood cells, and platelets.
This hemophagocytic activity primarily occurs within the reticuloendothelial system, most notably the bone marrow, spleen, liver, and lymph nodes, explaining the classic clinical findings of severe cytopenias and hepatosplenomegaly.
Classification of Hemophagocytic Lymphohistiocytosis (HLH)
For diagnostic and management purposes, Hemophagocytic Lymphohistiocytosis (HLH) is broadly divided into two categories, though the terminal pathways are identical:
- Primary (Familial) HLH (FHL): Driven by inherited genetic mutations affecting the cytotoxic pathway, classically presenting in infancy or early childhood.
- Secondary (Acquired) HLH (sHLH): Triggered by an underlying external event, most commonly severe infections (like Epstein-Barr Virus), malignancies (particularly T-cell or NK-cell lymphomas), or autoimmune/rheumatological conditions (often termed Macrophage Activation Syndrome or MAS).
Etiology and Causes of HLH
HLH is traditionally divided into two main etiological categories: Primary (Familial) HLH and Secondary (Acquired) HLH.
Increasingly, hematologists view these not as two distinct bins, but as a spectrum. A patient may have a subtle, heterozygous genetic predisposition that remains silent until a massive external trigger pushes their immune system over the edge.
Primary (Familial) HLH (FHL)
Primary Hemophagocytic Lymphohistiocytosis (HLH) is driven by underlying genetic mutations. It typically presents in infancy or early childhood (often within the first year of life), though adult-onset cases are increasingly recognized due to better genetic sequencing.
The genetic defects in FHL almost exclusively involve the perforin-dependent cytotoxic pathway. When a cytotoxic T-cell or Natural Killer (NK) cell recognizes an infected cell, it must form an immunological synapse, traffic lytic granules to the surface, fuse those granules with the cell membrane, and release perforin and granzymes to induce apoptosis. FHL mutations break specific steps in this assembly line.
Classic FHL Genetic Subtypes
- FHL-1: Gene defect unknown, mapped to chromosome 9.
- FHL-2 (PRF1 mutation): Defect in Perforin itself. Without perforin, pores cannot be formed in the target cell membrane, rendering granzymes useless.
- FHL-3 (UNC13D mutation): Defect in Munc13-4, a protein essential for the priming and fusion of lytic granules at the immunological synapse.
- FHL-4 (STX11 mutation): Defect in Syntaxin-11, a SNARE protein involved in intracellular membrane trafficking and granule exocytosis.
- FHL-5 (STXBP2 mutation): Defect in Munc18-2, which binds to syntaxin-11 and is crucial for granule exocytosis.
HLH Associated with Immunodeficiency Syndromes
Primary Hemophagocytic Lymphohistiocytosis (HLH) can also manifest as part of broader, known genetic immunodeficiencies.
- Chediak-Higashi Syndrome (LYST mutation): Look for partial albinism, giant granules in neutrophils, and peripheral neuropathy.
- Griscelli Syndrome Type 2 (RAB27A mutation): Look for partial albinism and immunodeficiency (without the giant granules).
- X-Linked Lymphoproliferative Disease (XLP): Typically triggered by an acute Epstein-Barr Virus (EBV) infection in young males. Includes XLP1 (SH2D1A / SAP mutation) and XLP2 (BIRC4 / XIAP mutation).
Secondary (Acquired) HLH (sHLH)
Secondary Hemophagocytic Lymphohistiocytosis (HLH) occurs when a strong immunological trigger sets off the hyperinflammatory cascade in a patient without a known familial Hemophagocytic Lymphohistiocytosis (HLH) mutation. This is the most common form seen in adult medicine and the intensive care unit.
The triggers for sHLH can be remembered by classifying them into three main buckets: Infectious, Malignant, and Autoimmune/Rheumatologic.
Infectious Triggers (The most common cause overall)
Viruses are the most notorious culprits, as they directly stimulate robust T-cell and macrophage responses.
- Viral: Epstein-Barr Virus (EBV) is the classic and most frequent infectious trigger. Others include Cytomegalovirus (CMV), HIV, Influenza, Dengue, and SARS-CoV-2.
- Bacterial: Tuberculosis, Brucellosis, severe sepsis, and atypical pathogens like Mycoplasma.
- Fungal & Parasitic: Histoplasmosis, Leishmaniasis, Malaria, and Babesiosis.
Malignancy-Triggered HLH
Malignancy-associated HLH (M-HLH) carries a particularly poor prognosis. The neoplastic cells themselves may secrete inflammatory cytokines, or the malignancy may disrupt normal immune regulation.
- Lymphomas: T-cell and NK-cell lymphomas are the most strongly associated. Diffuse Large B-Cell Lymphoma (DLBCL) and Hodgkin lymphoma can also be triggers.
- Leukemias: Acute lymphoblastic leukemia (ALL) and acute myeloid leukemia (AML).
Rheumatologic/Autoimmune Triggers (Macrophage Activation Syndrome - MAS)
When secondary Hemophagocytic Lymphohistiocytosis (HLH) is triggered by an underlying autoimmune or autoinflammatory disorder, it is specifically termed Macrophage Activation Syndrome (MAS).
- Systemic Juvenile Idiopathic Arthritis (sJIA) in pediatric populations.
- Adult-Onset Still's Disease (AOSD).
- Systemic Lupus Erythematosus (SLE) and Kawasaki disease.
Pathophysiology
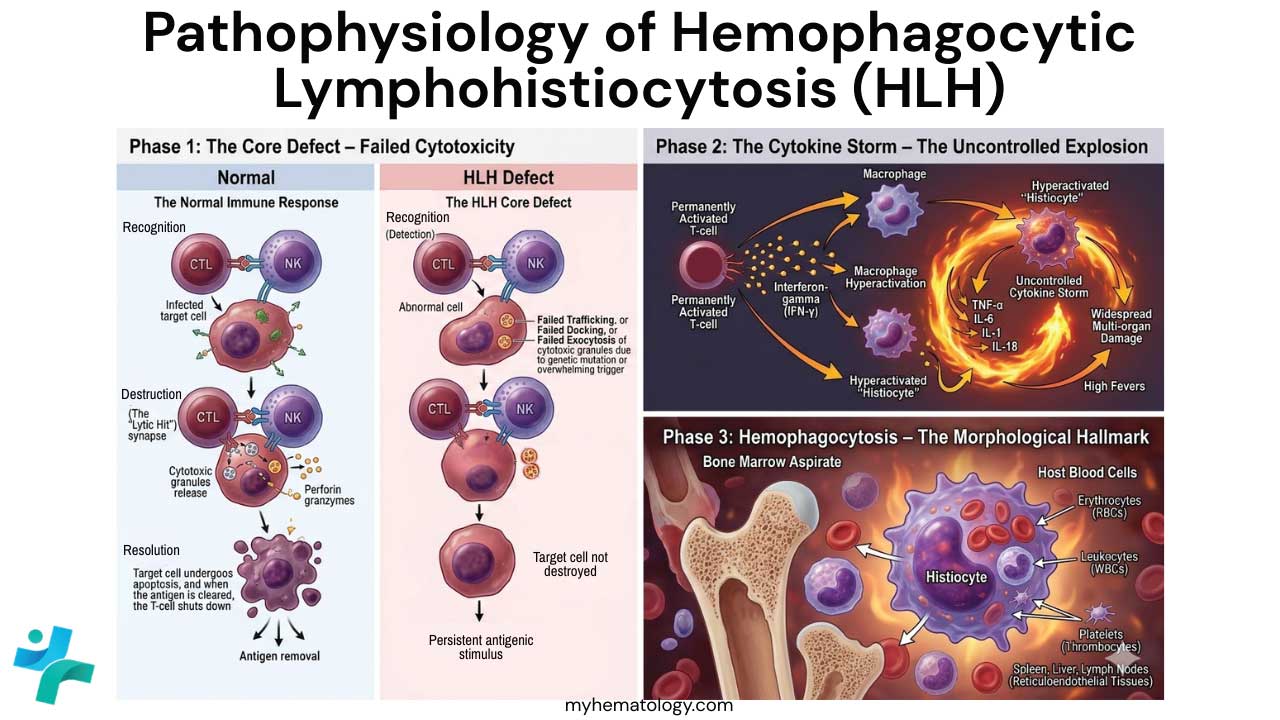
The Normal Immune Response
In a healthy individual, the immune system follows a regulated "Check-and-Balance" cycle to eliminate threats without damaging the host.
- Search (Recognition): An Antigen-Presenting Cell (APC) detects a threat (e.g., a virus-infected cell or a malignant cell). It presents the antigen to CD8+ Cytotoxic T-Lymphocytes (CTLs) and Natural Killer (NK) cells.
- Destroy (The Lytic Hit): The CTL or NK cell forms an "immunological synapse" with the target cell. It then fires its "weapons":
- Perforin: Puncture-forming proteins that create holes in the target cell membrane.
- Granzymes: Proteases that enter through those holes to trigger programmed cell death (apoptosis).
- Terminate (Resolution): Once the target cell is destroyed, the source of the antigen is gone. The T-cells stop receiving stimulatory signals, they undergo apoptosis or become memory cells, and the inflammatory response shuts down.
The HLH Pathophysiology
In Hemophagocytic Lymphohistiocytosis (HLH), the "Destroy" phase is mechanically broken, which prevents the "Terminate" phase from ever occurring.
- Defective Perforin-Mediated Cytotoxicity: Whether due to a genetic mutation (Primary HLH) or an overwhelming external trigger (Secondary HLH), the CTLs and NK cells are "disarmed." They may reach the target cell and recognize it, but they cannot deliver the lethal blow e.g. the perforin might be missing (FHL-2) or the lytic granules might be unable to move toward the cell surface or fuse with the membrane (FHL-3, 4, and 5).
- Failure to Clear the Antigen: Because the CTLs cannot kill the target cell, the antigenic stimulus persists. The T-cell remains "locked" to the target cell in a state of permanent activation. In a desperate attempt to clear the trigger, the T-cell begins to secrete massive, pathological amounts of Interferon-gamma (IFN-γ).
- The Recruitment of Macrophages: IFN-γ is the primary signal that activates macrophages. In Hemophagocytic Lymphohistiocytosis (HLH), the flood of IFN-γ recruits an army of macrophages and hyper-activates them. Unlike normal macrophages, these become "pathological histiocytes."
- The Cytokine Storm: These hyper-activated macrophages begin their own massive secretory phase, releasing a "storm" of pro-inflammatory cytokines:
- IL-6, IL-1b, and TNF-α: These drive the unremitting high fevers and systemic "wasting" appearance of the patient.
- High Ferritin: Driven by these cytokines, the body overproduces ferritin and fails to clear it, leading to the hallmark hyperferritinemia.
- Hemophagocytosis: Finally, these macrophages lose their ability to distinguish between the initial trigger and the host’s own healthy cells. They begin to phagocytose (eat) the patient’s own erythrocytes, leukocytes, and platelets within the bone marrow and lymphoid organs. This leads to the characteristic cytopenias (anemia, thrombocytopenia, neutropenia) seen on a peripheral blood film.
- Systematic Organ Destruction: The "Cytokine Storm" doesn't stay localized. It travels through the blood, leading to:
- Liver: Infiltration and cytokine damage (Elevated LFTs, low fibrinogen).
- Spleen: Infiltration and engorgement (Splenomegaly).
- Endothelium: Capillary leak and DIC (Disseminated Intravascular Coagulation).
- CNS: Microglial activation and inflammation (Neurological symptoms).
Clinical Manifestations
The clinical presentation is often non-specific and frequently mirrors severe sepsis or multi-organ failure. A high index of suspicion is required when a patient with "sepsis" does not respond to standard antibiotic therapy.
The clinical signs are a direct result of the "cytokine storm" and the infiltration of hyperactivated macrophages into various organs.
- Unremitting Fever: Almost all patients (>90%) present with a high-grade, persistent fever driven by high circulating levels of pyrogenic cytokines, particularly IL-1, IL-6, and TNF-α. Unlike typical infections, this fever often does not follow a "spike and recovery" pattern but remains stubbornly high.
- Organomegaly (Reticuloendothelial Infiltration):
- Splenomegaly: Found in the vast majority of cases. The spleen becomes engorged with activated macrophages and lymphocytes.
- Hepatomegaly & Liver Dysfunction: The liver is a primary target for infiltration. This manifests as jaundice, palpable enlargement, and clinically as a "pseudo-acute liver failure" (elevated transaminases and conjugated hyperbilirubinemia).
- Neurological Involvement (CNS HLH): This is a critical area as CNS involvement carries a much poorer prognosis and requires more aggressive therapy (like intrathecal chemotherapy). Symptoms include altered mental status, irritability (especially in infants), seizures, meningismus, or cranial nerve palsies. Neurological signs may be subtle initially, but their presence suggests that the inflammatory process has crossed the blood-brain barrier.
- Dermatological Manifestations: Rashes are common but highly variable as they can be maculopapular, morbilliform, or even resemble erythroderma. Petechiae and purpura may appear, not due to the rash itself, but as a secondary effect of severe thrombocytopenia and DIC.
- The Effects of Cytopenias: As the macrophages "eat" the blood lines in the bone marrow, the physical consequences become apparent:
- Pallor: From severe anemia.
- Bleeding/Bruising: From profound thrombocytopenia.
- Increased Infection Risk: From neutropenia (though this is often masked by the existing inflammatory state).
Laboratory Investigations of HLH
Diagnostic Criteria
To meet the clinical diagnosis of Hemophagocytic Lymphohistiocytosis (HLH) (in the absence of a known genetic defect), a patient must fulfill 5 out of 8 of the following criteria.
| Parameter | Threshold | Pathophysiological Context |
| Cytopenias | ≥ 2 lineages (Hb < 90 g/L, Plt < 100 x 109/L, Neut < 1.0 x 109/L) | Result of hemophagocytosis and high TNF-α levels. |
| Ferritin | ≥ 500 ng/mL | A marker of extreme macrophage activation. |
| Triglycerides | ≥ 3.0 mmol/L (or 265 mg/dL) | High TNF-α inhibits lipoprotein lipase. |
| Fibrinogen | ≤ 1.5 g/L | Consumed due to hyperinflammation and plasminogen activator release. |
| sCD25 (sIL-2R) | ≥ 2400 U/mL | Reflects massive T-cell activation. |
| NK-Cell Activity | Low or absent | Indicates a defect in the "kill" signal of the immune system. |
| Hemophagocytosis | Positive in Bone Marrow/Spleen/LN | Morphological evidence of macrophages "eating" blood cells. |
| Fever | ≥ 38.5°C | Driven by IL-1, IL-6, and TNF-α |
The "Ferritin Rule"
While the threshold is ≥ 500 ng/mL, clinicians look for much higher values.
- Ferritin > 10,000 ng/mL: Highly specific for Hemophagocytic Lymphohistiocytosis (HLH) in pediatric populations (approx. 90% sensitive and 96% specific).
- Prognostic Value: A failure of ferritin levels to drop by at least 50% within the first 48 - 72 hours of treatment is a strong predictor of poor outcomes.
Specialized & Functional Testing
For patients suspected of Primary (Genetic) HLH, specialized flow cytometry is used to identify specific protein defects before genetic sequencing returns.
- Perforin Expression: Low levels suggest mutations in the PRF1 gene (FHL2).
- CD107a Degranulation Assay: Measures the ability of NK cells to release cytotoxic granules. If low, it suggests defects in the "machinery" of exocytosis (e.g., UNC13D, STX11, STXBP2).
- Munc13-4 Protein: Specific flow testing for the protein product of UNC13D.
Emerging Biomarkers
Recent advancements have introduced markers that help differentiate Hemophagocytic Lymphohistiocytosis (HLH) from other "cytokine storm" mimics like Macrophage Activation Syndrome (MAS) or Sepsis:
- CXCL9 (Interferon-γ Surrogate): Since IFN-γ has a very short half-life, CXCL9 is a more stable marker of the IFN-γ pathway. Levels > 16,100 pg/mL are now recognized as independent predictors of high mortality.
- IL-18 / CXCL9 Ratio:
- High IL-18 + Lower CXCL9: Points toward MAS (associated with Still’s disease).
- High CXCL9 + Lower IL-18: Points toward Primary HLH or EBV-HLH.
- IL-18 Thresholds: In Still's-related MAS, IL-18 often exceeds 24,000 pg/mL, whereas it is significantly lower in other forms of HLH.
Etiological Investigations
Once Hemophagocytic Lymphohistiocytosis (HLH) is confirmed, you must find the "Trigger":
- Viral PCRs: Mandatory screening for EBV, CMV, and HIV. EBV-associated Hemophagocytic Lymphohistiocytosis (HLH) is a medical emergency requiring specific B-cell depletion (Rituximab).
- Malignancy Screen: PET/CT or repeat Bone Marrow biopsies to rule out Lymphoma (the most common adult trigger).
- Autoimmune Markers: ANA, dsDNA, and C3/C4 to screen for SLE-associated MAS.
HScore
The HLH-2004 criteria were originally designed for pediatric patients with genetic (Primary) HLH and often perform poorly in adult populations with Secondary HLH.
The HScore is a weighted scoring system that estimates the individual probability of a patient having HLH. Unlike the "binary" HLH-2004 criteria (where you either meet 5/8 or you don't), the HScore provides a probability percentage.
The 9 Parameters of the HScore
Each parameter is assigned a specific number of points based on its severity.
| Parameter | Criteria / Thresholds | Points |
| Known Immunosuppression | No / Yes (e.g., HIV, post-transplant, steroids) | 0 / 18 |
| Temperature (°C) | <38.4 / 38.4–39.4 / >39.4 | 0 / 33 / 49 |
| Organomegaly | None / Hepatomegaly or Splenomegaly / Both | 0 / 38 / 73 |
| Number of Cytopenias | 1 lineage / 2 lineages / 3 lineages | 0 / 34 / 58 |
| Triglycerides (mmol/L) | <1.5 / 1.5–4.0 / >4.0 | 0 / 44 / 64 |
| Fibrinogen (g/L) | >2.5 / <2.5 | 0 / 30 |
| Ferritin (ng/mL) | <2,000 / 2,000–6,000 / >6,000 | 0 / 35 / 50 |
| AST (U/L) | <30 / >30 | 0 / 19 |
| Hemophagocytosis (BM) | No / Yes | 0 / 35 |
Note: Cytopenia is defined as Hemoglobin <9.2 g/dL, Leukocytes <4,000/µL, and Platelets <110,000/µL.
Interpreting the Score
The total points are summed to determine the probability of Hemophagocytic Lymphohistiocytosis (HLH). While there isn't a single "cut-off" that is 100% accurate, the following thresholds are generally used in clinical practice:
- Total Score of 169: Corresponds to a 60–70% probability of Hemophagocytic Lymphohistiocytosis (HLH). This is often the threshold where clinicians begin to consider treatment seriously.
- Total Score of 200: Corresponds to a >90% probability of Hemophagocytic Lymphohistiocytosis (HLH).
- Total Score of 250: Corresponds to a >99% probability of Hemophagocytic Lymphohistiocytosis (HLH).
Differential Diagnosis
Because the hallmark signs of Hemophagocytic Lymphohistiocytosis (HLH) - fever, cytopenias, and organ dysfunction - are found in many critical illnesses, Hemophagocytic Lymphohistiocytosis (HLH) is often referred to as a "great masquerader."
Sepsis and Septic Shock
This is the most common and difficult distinction to make. Both present with high fever, tachycardia, hypotension, and multi-organ failure. Both feature systemic inflammatory response syndrome (SIRS).
- Fibrinogen: In sepsis (an acute-phase state), fibrinogen is typically high. In Hemophagocytic Lymphohistiocytosis (HLH), it is frequently low due to consumption.
- Ferritin: While ferritin can rise in sepsis, it rarely exceeds the 5,000–10,000 ng/mL range seen in Hemophagocytic Lymphohistiocytosis (HLH).
- Splenomegaly: Common in Hemophagocytic Lymphohistiocytosis (HLH), but rare in straightforward bacterial sepsis.
- Antibiotic Response: Hemophagocytic Lymphohistiocytosis (HLH) will not improve with broad-spectrum antibiotics alone.
Thrombotic Microangiopathies (TTP/HUS)
Thrombotic Thrombocytopenic Purpura (TTP) and Hemolytic Uremic Syndrome (HUS) can mimic the "hematological collapse" seen in HLH. Fever, thrombocytopenia, and multi-organ (especially renal or CNS) involvement.
- Schistocytes: TTP/HUS will show significant red cell fragmentation (schistocytes) on a peripheral film. Hemophagocytic Lymphohistiocytosis (HLH) usually shows a "clean" film, perhaps with some signs of hemophagocytosis in the bone marrow but not schistocytes.
- Coagulation: TTP generally has normal coagulation studies (PT/PTT/Fibrinogen). Hemophagocytic Lymphohistiocytosis (HLH) often presents with a DIC-like picture (prolonged times, low fibrinogen).
Macrophage Activation Syndrome (MAS)
MAS is actually a form of secondary Hemophagocytic Lymphohistiocytosis (HLH), but it is categorized separately in rheumatology. It is triggered by autoimmune diseases like Systemic Juvenile Idiopathic Arthritis (sJIA) or Lupus (SLE). Clinical symptoms are identical.
- Underlying Diagnosis: A known history of Still’s disease or SLE.
- IL-18 Levels: MAS typically features much higher levels of Interleukin-18 (>20,000 pg/mL) compared to primary HLH.
- ESR Paradox: In MAS, the ESR (Erythrocyte Sedimentation Rate) may suddenly drop despite worsening inflammation because the fibrinogen is being consumed.
Acute Leukemia and Lymphoma
Malignancies can both trigger Hemophagocytic Lymphohistiocytosis (HLH) and mimic it, particularly T-cell and NK-cell lymphomas. Clinical symptoms for both are fever, cytopenias, and hepatosplenomegaly.
- Bone Marrow Aspirate: In leukemia, you will see a population of malignant blasts. In Hemophagocytic Lymphohistiocytosis (HLH), the marrow might be hypocellular or normocellular, but it will show activated macrophages engulfing healthy cells.
- Lymph Node Biopsy: Necessary if lymphoma is suspected.
Kawasaki Disease
In pediatric populations, severe Kawasaki Disease can look very similar to HLH, especially in cases of "Incomplete Kawasaki." Both have prolonged high fever, rash, and lymphadenopathy.
- Platelet Count: KD is typically characterized by thrombocytosis (high platelets) after the first week. HLH is characterized by thrombocytopenia (low platelets).
- Ferritin: KD does not typically reach the extreme hyperferritinemia seen in HLH.
Summary of Differential Diagnosis
| Feature | HLH | Sepsis | TTP | MAS |
| Ferritin | Extremely High | Moderate | Mildly High | Extremely High |
| Fibrinogen | Low | High | Normal | Low |
| Splenomegaly | Present | Rare | Absent | Present |
| Schistocytes | Absent | Absent | Present | Absent |
| Triglycerides | High | Normal | Normal | High |
Treatment and Management of HLH
The management of Hemophagocytic Lymphohistiocytosis (HLH) is a medical emergency. The therapeutic goal is twofold: first, to suppress the life-threatening hyperinflammation, and second, to treat the underlying trigger.
Because the mortality rate for untreated Hemophagocytic Lymphohistiocytosis (HLH) is nearly 100%, treatment must often be initiated based on clinical suspicion before all diagnostic results (like genetic testing) return.
Immediate Stabilization and Supportive Care
Before specific immunosuppression begins, the patient requires intensive supportive management:
- Blood Product Support: Aggressive transfusion of platelets, packed red cells, and cryoprecipitate (to maintain fibrinogen levels > 1.5 g/L).
- Infection Prophylaxis: Due to the upcoming intense immunosuppression, patients need protection against opportunistic infections (e.g., Pneumocystis jirovecii prophylaxis and antifungal coverage).
- Organ Support: Many patients require ICU-level care for mechanical ventilation or continuous renal replacement therapy (CRRT).
The Standard Induction: The HLH-94 and HLH-2004 Protocols
The "gold standard" for treating both primary and severe secondary HLH is the HLH-94 protocol (with slight modifications from the HLH-2004 trial). This intensive 8-week induction phase aims to "calm the storm" using three main agents:
- Etoposide (VP-16): A chemotherapy agent that is uniquely effective in Hemophagocytic Lymphohistiocytosis (HLH) is a medical emergency. The therapeutic goal is twofold: first, to suppress the life-threatenin because it selectively induces apoptosis in activated T-cells and stops the production of inflammatory cytokines.
- Dexamethasone: A potent corticosteroid that suppresses macrophage activation and, crucially, crosses the blood-brain barrier to treat Central Nervous System (CNS) involvement.
- Cyclosporine A: A calcineurin inhibitor added to maintain T-cell suppression (usually introduced after the initial induction).
Trigger-Specific Management
In Secondary Hemophagocytic Lymphohistiocytosis (HLH), treating the trigger is just as important as suppressing the inflammation:
- Viral HLH (especially EBV): Rituximab (anti-CD20) is added to deplete B-cells, which are the primary reservoir for the Epstein-Barr Virus. This "removes the fuel" for the T-cell activation.
- Malignancy-Associated HLH: Treatment must transition to malignancy-specific chemotherapy as soon as the initial hyperinflammation is stabilized.
- Autoimmune (MAS): Often responds to high-dose steroids and Anakinra (an IL-1 receptor antagonist) rather than etoposide.
Salvage and Targeted Therapies
For patients who do not respond to the standard HLH-94 protocol, newer biological therapies have revolutionized care:
- Emapalumab: A monoclonal antibody that binds directly to Interferon-gamma (IFN-γ), neutralizing the primary driver of the cytokine storm. It is now FDA-approved for primary Hemophagocytic Lymphohistiocytosis (HLH) that is refractory or recurrent.
- Anakinra / Canakinumab: Targets Interleukin-1 (IL-1), particularly useful in Macrophage Activation Syndrome (MAS).
- Tocilizumab: Targets Interleukin-6 (IL-6); though used in COVID-19 cytokine storms, its use in classic Hemophagocytic Lymphohistiocytosis (HLH) is more cautious as IL-6 is only one part of the multi-cytokine storm.
- JAK Inhibitors (e.g., Ruxolitinib): These oral medications block the signaling pathways of multiple cytokines simultaneously and are showing great promise in clinical trials for secondary Hemophagocytic Lymphohistiocytosis (HLH).
Allogeneic HSCT
For patients with Primary (Genetic) HLH, or those with relapsed/refractory Secondary Hemophagocytic Lymphohistiocytosis (HLH), the only definitive cure is an Allogeneic Hematopoietic Stem Cell Transplant (HSCT).
Frequently Asked Questions (FAQs)
Is HLH considered a hematologic malignancy?
No. Hemophagocytic Lymphohistiocytosis (HLH) is a hyperinflammatory syndrome, not a cancer. However, it is closely linked to oncology because malignancies, specifically T-cell and NK-cell lymphomas, are notorious triggers for Secondary HLH. Additionally, the treatments used to suppress the immune system in Hemophagocytic Lymphohistiocytosis (HLH) (like etoposide) are traditional chemotherapy agents.
Why does ferritin reach such extreme levels in HLH compared to normal sepsis?
Ferritin is an acute-phase reactant, so it rises in any infection. However, macrophages are the primary storage sites for ferritin. In Hemophagocytic Lymphohistiocytosis (HLH), the intense, pathological hyperactivation of these macrophages by Interferon-gamma (IFN-γ), combined with the active engulfment of erythrocytes (which contain iron), leads to a massive synthesis and release of ferritin into the bloodstream. Levels exceeding 10,000 ng/mL are highly suggestive of HLH.
What is the difference between Macrophage Activation Syndrome (MAS) and HLH?
MAS is pathophysiologically identical to Secondary HLH. The term MAS is specifically reserved for cases of HLH that are triggered by underlying autoimmune or rheumatologic conditions, most commonly Systemic Juvenile Idiopathic Arthritis (sJIA) or Adult-Onset Still's Disease (AOSD).
Can a patient have both a genetic mutation AND an infectious trigger?
Yes, and this is a classic board-exam concept. The line between Primary and Secondary HLH is blurring. A patient may have a heterozygous or "hypomorphic" (partial loss of function) mutation in a perforin gene. This genetic defect might remain completely silent until a massive external trigger, like an acute Epstein-Barr Virus (EBV) infection, stresses the immune system enough to push it into a full cytokine storm.
Why are antibiotics ineffective in treating the fever of HLH?
The unremitting fever in Hemophagocytic Lymphohistiocytosis (HLH) is not caused by bacterial pyrogens; it is an endogenous fever driven by the continuous, massive secretion of pro-inflammatory cytokines (IL-1, IL-6, and TNF-α) from hyperactivated macrophages. Until the macrophage activation is shut down with immunosuppression (like dexamethasone or etoposide), the fever will persist.
Glossary of Key Medical Terms
- Cytokine Storm: A severe, systemic immune reaction caused by the rapid, uncontrolled release of pro-inflammatory cytokines into the bloodstream, driving multi-organ failure.
- Etoposide (VP-16): A chemotherapeutic drug functioning as a topoisomerase II inhibitor. In the HLH-94 protocol, it is uniquely effective because it selectively induces apoptosis in hyperactivated T-cells.
- Granzymes: Serine proteases stored within the cytoplasmic granules of cytotoxic T-cells and Natural Killer (NK) cells. They enter target cells through pores to trigger programmed cell death (apoptosis).
- Hemophagocytosis: The defining morphological feature of Hemophagocytic Lymphohistiocytosis (HLH), characterized by the active engulfment (phagocytosis) and destruction of host hematopoietic cells (red blood cells, white blood cells, and platelets) by pathologically activated macrophages in the bone marrow and reticuloendothelial system.
- Histiocytes: A stationary phagocytic cell present in connective tissues. In the context of HLH, the term refers to the tissue-resident macrophages that become aggressively and inappropriately activated.
- Hyperferritinemia: Extremely elevated levels of ferritin in the blood. A key laboratory marker for HLH, often reaching levels > 10,000 ng/mL.
- Interferon-gamma: A cytokine critically involved in innate and adaptive immunity against viral and intracellular bacterial infections. In HLH, massive secretion by T-cells drives the hyperactivation of macrophages.
- Macrophage Activation Syndrome (MAS): A life-threatening complication of rheumatic diseases (e.g., Still's disease, systemic juvenile idiopathic arthritis), classified pathophysiologically as a form of secondary (acquired) HLH.
- Perforin: A cytolytic protein found in the granules of cytotoxic T-cells and NK cells. Upon degranulation, it inserts itself into the target cell's membrane, forming pores that allow granzymes to enter.
- Primary HLH (Familial HLH or FHL): Hemophagocytic Lymphohistiocytosis (HLH) caused by inherited, autosomal recessive genetic mutations affecting the perforin-dependent cytotoxic pathway.
- Secondary HLH (Acquired HLH): Hemophagocytic Lymphohistiocytosis (HLH) occurring in patients without a known underlying genetic defect, triggered by strong immunological stimuli such as severe infections, malignancies, or autoimmune diseases.
Disclaimer: This article is intended for informational purposes only and is specifically targeted towards medical students. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Konkol S, Killeen RB, Rai M. Hemophagocytic Lymphohistiocytosis. [Updated 2025 May 3]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2026 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK557776/
- Naymagon L. (2021). Can we truly diagnose adult secondary hemophagocytic lymphohistiocytosis (HLH)? A critical review of current paradigms. Pathology, research and practice, 218, 153321. https://doi.org/10.1016/j.prp.2020.153321
- Mirza, M., Zafar, M., Nahas, J., Arshad, W., Abbas, A., & Tauseef, A. (2021). Hemophagocytic lymph histiocytosis (HLH): etiologies, pathogenesis, treatment, and outcomes in critically ill patients: a review article and literature to review. Journal of community hospital internal medicine perspectives, 11(5), 639–645. https://doi.org/10.1080/20009666.2021.1954783
- Ponnatt, T. S., Lilley, C. M., & Mirza, K. M. (2022). Hemophagocytic Lymphohistiocytosis. Archives of pathology & laboratory medicine, 146(4), 507–519. https://doi.org/10.5858/arpa.2020-0802-RA
- Knaak, C., Nyvlt, P., Schuster, F. S., Spies, C., Heeren, P., Schenk, T., Balzer, F., La Rosée, P., Janka, G., Brunkhorst, F. M., Keh, D., & Lachmann, G. (2020). Hemophagocytic lymphohistiocytosis in critically ill patients: diagnostic reliability of HLH-2004 criteria and HScore. Critical care (London, England), 24(1), 244. https://doi.org/10.1186/s13054-020-02941-3
- Hayden, A., Park, S., Giustini, D., Lee, A. Y., & Chen, L. Y. (2016). Hemophagocytic syndromes (HPSs) including hemophagocytic lymphohistiocytosis (HLH) in adults: A systematic scoping review. Blood reviews, 30(6), 411–420. https://doi.org/10.1016/j.blre.2016.05.001
- Zoref-Lorenz, A., Witzig, T. E., Cerhan, J. R., & Jordan, M. B. (2025). Malignancy-associated HLH: mechanisms, diagnosis, and treatment of a severe hyperinflammatory syndrome. Leukemia & lymphoma, 66(4), 628–636. https://doi.org/10.1080/10428194.2024.2436037