Key Takeaways
Polycythemia means an abnormally high red blood cell count, reflected by raised hemoglobin and hematocrit on a full blood count.
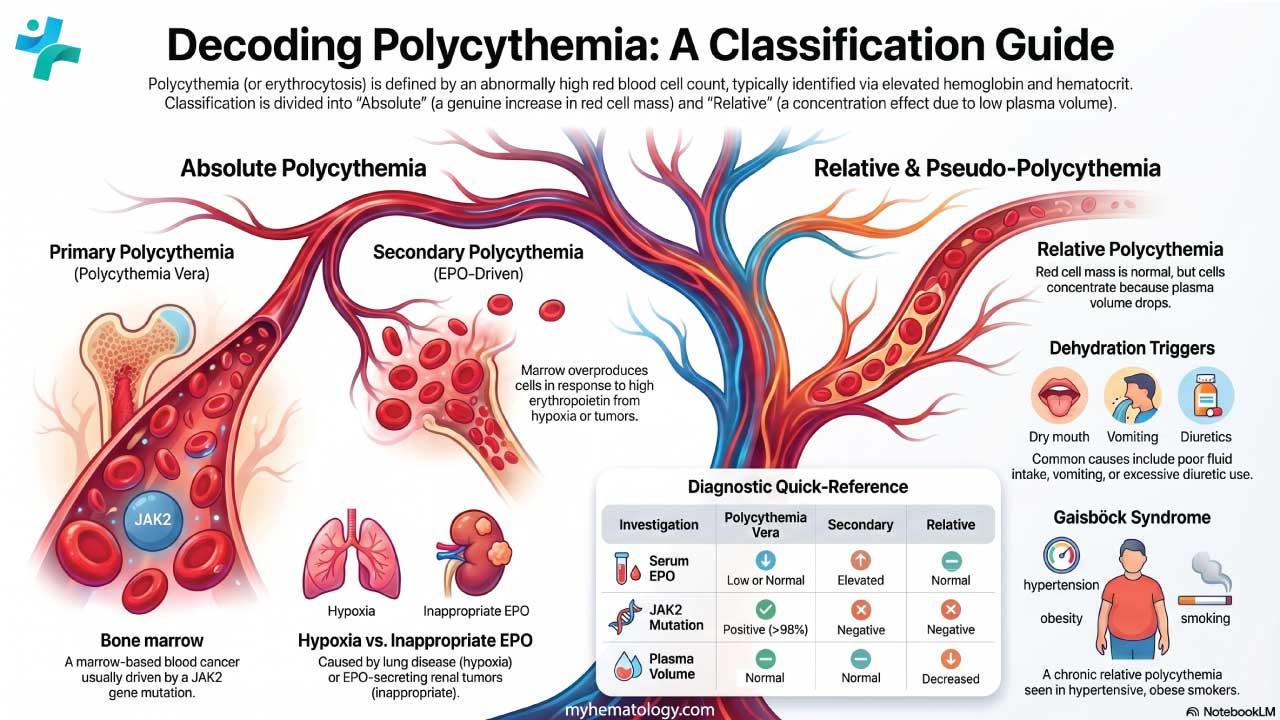
- Classification ▾: There are three types; primary (polycythemia vera, driven by a JAK2 mutation), secondary (driven by high erythropoietin from low oxygen or tumors), and relative (a fake rise caused by low plasma volume, usually dehydration).
- Symptoms & Signs ▾: These include headache, dizziness, fatigue, pruritus, plethora, splenomegaly, and increased risk of thrombosis and hemorrhage. Secondary polycythemia may have symptoms of the underlying cause.
- Laboratory Investigations ▾: Polycythemia vera is diagnosed using WHO 2022 criteria combining elevated Hb/Hct, JAK2 testing, low erythropoietin, and bone marrow morphology.
- Treatment & Management ▾: The hematocrit target in PV is <45% for both sexes, based on the CYTO-PV trial, achieved through phlebotomy, low-dose aspirin, and cytoreductive drugs [1].
*Click ▾ for more information
Introduction
Polycythemia is one of those findings on a blood report that looks simple but carries real weight. The hemoglobin is high. The hematocrit is high. What does it actually mean?
Polycythemia, also called erythrocytosis, is an abnormally high red blood cell (RBC) count. Because hemoglobin sits inside red cells, and hematocrit measures the share of blood made up of red cells, all three values rise together. The cause matters enormously, because polycythemia can be a harmless lab quirk, a sign of an underlying lung or kidney problem, or the first clue to a blood cancer called polycythemia vera.
Spotting it early lets clinicians find the cause, prevent clots, and treat any tumor or hypoxic disease driving it. This article walks through how polycythemia is classified, why it happens, what it looks like in patients, how it is diagnosed, and how the treatment landscape has shifted in 2024–2026.

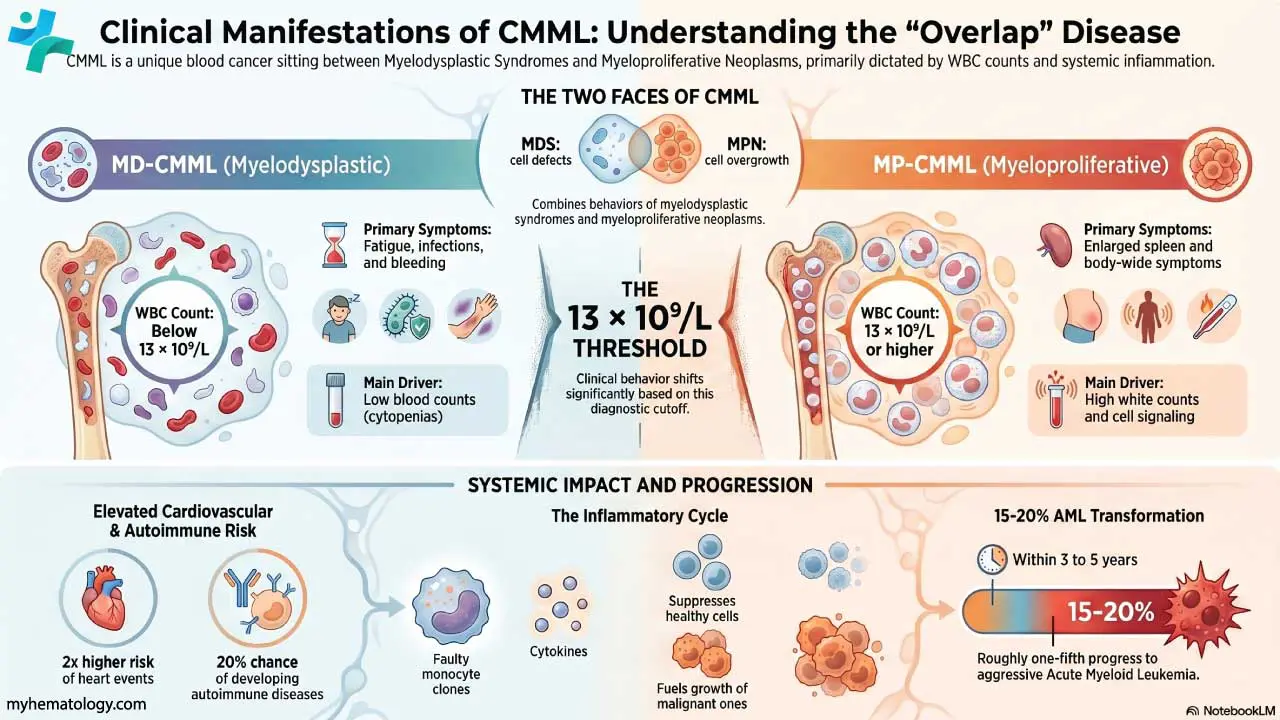
Classification of Polycythemia
Polycythemia splits into two main groups based on whether the red cell mass is genuinely raised.
Absolute polycythemia. The body really does have too many red cells. This group divides further into primary and secondary forms. Primary means the problem starts in the bone marrow itself. The classic example is polycythemia vera (PV), a myeloproliferative neoplasm almost always linked to a JAK2 mutation [2,3]. Secondary means the marrow is responding normally to an abnormal signal — usually too much erythropoietin (EPO), the hormone that drives red cell production.
Relative polycythemia (pseudopolycythemia). The red cell mass is normal, but the plasma volume has dropped. The cells are simply more concentrated. Dehydration is the most common cause. Once fluid is replaced, the blood counts normalize.
Causes and Pathophysiology
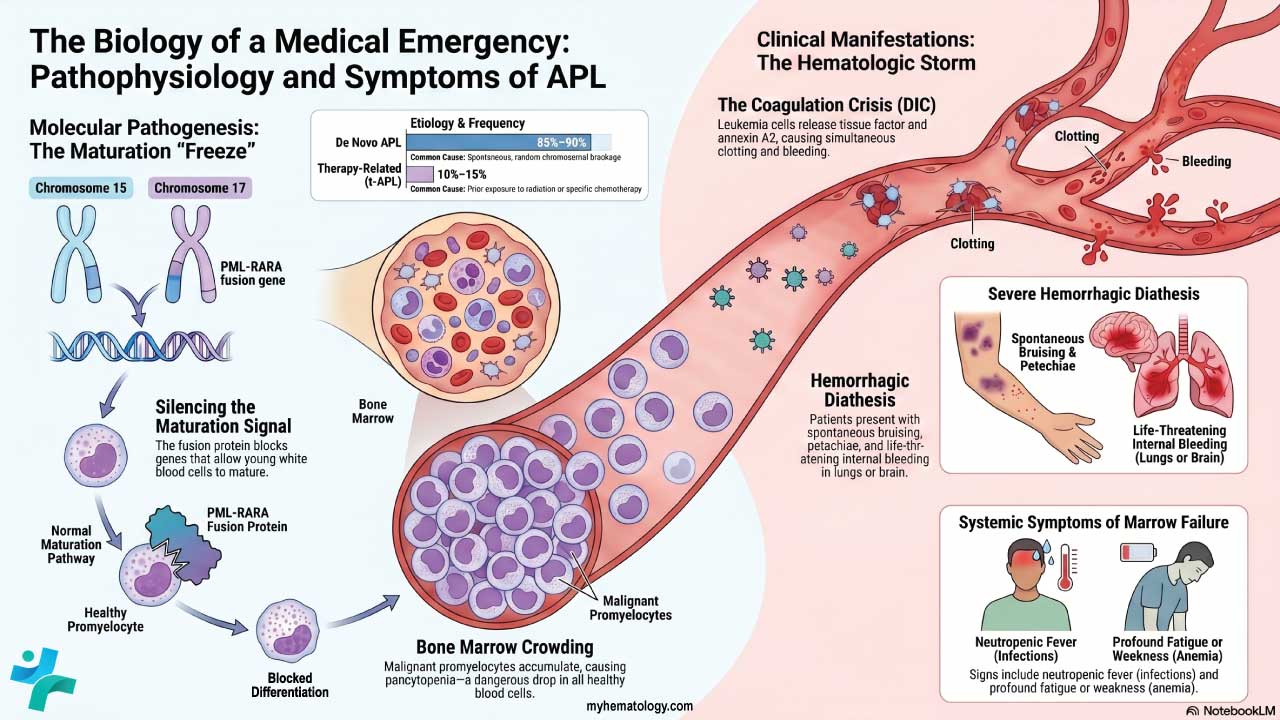
Polycythemia vera
PV is driven by a somatic mutation in the JAK2 gene, most often JAK2 V617F, present in about 95% of cases. A smaller group (around 3%) carry JAK2 exon 12 mutations, and together these two account for more than 98% of patients [7]. The mutation switches on JAK2 protein signaling without needing EPO, so erythroid progenitors keep multiplying.
The hallmark is clonal hematopoiesis where a single mutated stem cell taking over blood production. Although red cells dominate, white cells and platelets often rise too. The extra red cells thicken the blood (hyperviscosity), which sets the stage for clotting and many of the symptoms below. EPO levels are low or normal in PV, because the bone marrow no longer needs the signal to keep producing.
Secondary polycythemia
Here the marrow is normal, but something is telling it to produce more red cells.
Hypoxia-driven (appropriate EPO rise):
- Living at high altitude
- Chronic lung disease such as COPD
- Obstructive sleep apnea
- Cyanotic congenital heart disease
- Rare high-affinity hemoglobins (e.g., Hemoglobin Chesapeake) that hold onto oxygen too tightly, tricking the kidney into sensing hypoxia
Inappropriate EPO rise (no real hypoxia):
- EPO-secreting tumors, most commonly renal cell carcinoma, also hepatocellular carcinoma, pheochromocytoma, and cerebellar hemangioblastoma
- Exogenous EPO (used clinically in chronic kidney disease, misused as a doping agent)
- Androgen therapy
Inherited (congenital) causes include mutations in oxygen-sensing genes such as VHL, EGLN1, and EPOR.
Relative polycythemia
Plasma volume falls and the cells concentrate. Common triggers: dehydration from poor intake, vomiting, diarrhea, sweating, or diuretics. Gaisböck syndrome is a chronic form seen in obese, hypertensive smokers. The total red cell mass stays normal, so correcting fluid balance fixes the lab values.
Signs and Symptoms
Symptoms come from two sources: thickened blood flowing poorly, and (in PV) the wider effects of a myeloproliferative disease.
General symptoms — headache, dizziness, fatigue, blurred or double vision, and night sweats. Aquagenic pruritus, an intense itching triggered by warm water, is a hallmark of PV and one of its most distressing features.
Signs of increased viscosity and volume — a ruddy, flushed face (plethora), engorged retinal veins on fundoscopy, enlarged spleen (splenomegaly), and high blood pressure.
Thrombosis is the most dangerous consequence. Patients with polycythemia, especially PV, face elevated risk of stroke, transient ischemic attack, myocardial infarction, deep vein thrombosis, pulmonary embolism, and Budd-Chiari syndrome (clotting in the hepatic veins). For caregivers, this is the risk that drives most of the treatment decisions.
Paradoxical bleeding can also occur in PV like nosebleeds, gum bleeding, easy bruising, gastrointestinal bleeding because platelet function is often abnormal even when platelet counts are high.
In secondary polycythemia, symptoms of the underlying disease usually dominate: chronic cough and shortness of breath in lung disease, cyanosis in cyanotic heart disease, loud snoring and daytime sleepiness in obstructive sleep apnea, flank pain or hematuria in renal cell carcinoma.
Laboratory Investigations
Diagnosis hinges on the full blood count, then targeted follow-up tests to identify the type of polycythemia and its cause.
Step 1: Confirm the picture
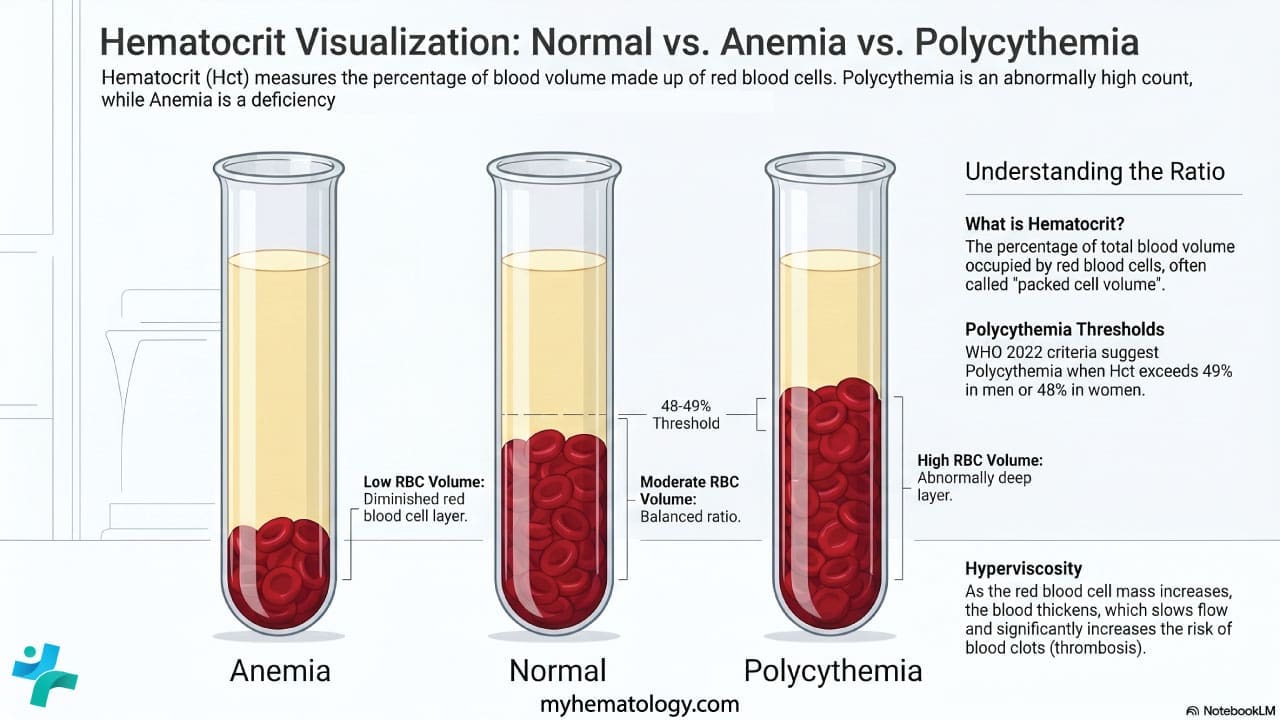
Using WHO 2022 thresholds, polycythemia is suggested by:
White cell and platelet counts may also be raised in PV, supporting its diagnosis as a multilineage myeloproliferative disease.
Masked PV
Some patients have Hb/Hct below these cutoffs due to concurrent iron deficiency or expanded plasma volume, yet possess a genuine expansion of red cell mass. If a patient presents with unexplained splanchnic vein thrombosis, intractable aquagenic pruritus, or a JAK2 mutation, a bone marrow biopsy is mandatory to evaluate for PV [8]
Step 2: Sort absolute from relative
Repeat the complete blood count after good hydration. If the values normalize, the polycythemia was relative. Direct measurement of red cell mass and plasma volume by isotope studies is the gold standard but is rarely used today.
Step 3: Sort primary from secondary
This is where the workup branches sharply.
- Serum erythropoietin (EPO). Low or normal EPO points to PV; high EPO points to a secondary cause.
- JAK2 mutation testing. Testing should be performed sequentially: peripheral blood is first screened for the JAK2 V617F mutation (~95% of cases). Only if this is negative should the more specialized test for JAK2 exon 12 mutations be ordered. A positive result strongly supports PV [7].
- Bone marrow biopsy. In PV, the marrow is hypercellular with hyperplasia of all three lineages and large, pleomorphic megakaryocytes. Reticulin fibrosis hints at progression toward myelofibrosis. Bone marrow examination is a major WHO 2022 diagnostic criterion [2,3].
- Arterial blood gas. A low PaO2 supports a hypoxia-driven secondary cause.
- Imaging. Renal ultrasound or CT of the abdomen looks for EPO-secreting tumors when secondary polycythemia is suspected.
- Genetic testing for VHL, EGLN1, EPOR, or high-affinity hemoglobin variants when congenital erythrocytosis is on the differential.
Other useful labs: iron studies (PV patients are often iron-deficient because of phlebotomy or marrow utilization), vitamin B12 and folate, liver and kidney function, uric acid (often raised in PV), and LDH..
Laboratory findings across the three types of polycythemia
| Investigation | Polycythemia vera | Secondary polycythemia | Relative polycythemia |
|---|---|---|---|
| Hb, Hct, RBC | Elevated | Elevated | Elevated |
| Red cell indices | Usually normocytic, normochromic | Usually normocytic, normochromic | Variable |
| WBC, platelets | Often elevated | Normal to mildly elevated | Normal |
| Serum EPO | Low or normal | Elevated | Normal |
| JAK2 mutation | Positive in >98% | Negative | Negative |
| Bone marrow | Hypercellular, trilineage hyperplasia | Erythroid hyperplasia | Normal |
| Arterial blood gas | Normal | Low PaO2 if hypoxic | Normal |
| Plasma volume | Normal | Normal | Decreased |
| Uric acid | Often elevated | Variable | Normal |
Treatment and Management
The goals are simple: lower the clot risk, ease symptoms, and treat the cause when possible. The methods differ sharply between the three types.
Polycythemia vera
Phlebotomy stays at the foundation of care. Removing 300–500 mL of blood at intervals brings the hematocrit down and keeps it there. The target is <45% for both men and women, based on the CYTO-PV trial, which showed significantly fewer cardiovascular deaths and major clots at this threshold [1].
Low-dose aspirin (typically 75–100 mg daily) is recommended for most patients to cut thrombotic risk, unless there is a clear bleeding contraindication. Additionally, aggressive management of modifiable cardiovascular risk factors (smoking, hypertension, diabetes) is critical, as these independently multiply the risk of major thrombotic events [9].
Cytoreductive therapy is added for high-risk patients (age >60, prior thrombosis, or poor control on phlebotomy alone). The landscape has shifted noticeably:
- Ropeginterferon alfa-2b (Besremi) is now the NCCN-preferred first-line cytoreductive drug for both low- and high-risk PV, regardless of treatment history [5]. The PROUD-PV/CONTINUATION-PV trials showed durable hematologic response and reduction in JAK2 allele burden over years of treatment.
- Hydroxyurea (hydroxycarbamide) remains widely used and effective, particularly in older patients. It blocks DNA synthesis and lowers all blood lineages.
- Ruxolitinib, a JAK1/JAK2 inhibitor, is the standard option when hydroxyurea fails or is not tolerated. The MAJIC-PV trial showed superior complete responses, around 40% reduction in thrombotic events, and molecular responses linked to event-free survival [4].
- Rusfertide, an investigational hepcidin mimetic, limits iron availability for red cell production. In the phase 2 REVIVE trial reported in 2024, it effectively controlled hematocrit and kept ~60% of patients phlebotomy-free through 52 weeks [6]. The confirmatory phase 3 VERIFY trial was subsequently launched to support regulatory submission.
- Busulfan and pipobroman are older alkylating agents used selectively in elderly patients but carry a higher long-term leukemogenic risk.
Symptom management addresses aquagenic pruritus (SSRIs such as paroxetine, antihistamines, interferon, or PUVA therapy), hyperuricemia (allopurinol if gout or kidney stones are a concern), and progression to post-polycythemic myelofibrosis (treated as primary myelofibrosis, sometimes with stem cell transplantation).

Secondary polycythemia
Here the priority is the underlying cause.
- Smoking cessation, oxygen therapy, and management of COPD or cyanotic heart disease for hypoxia-driven cases
- CPAP for obstructive sleep apnea
- Surgical resection of EPO-secreting tumors
- Stopping exogenous EPO or androgens where possible
- Phlebotomy may be used to relieve symptoms while the underlying disease is being treated, but it is not a long-term solution on its own
Relative polycythemia
Restore plasma volume. Increase oral fluids, give intravenous fluids when appropriate, and adjust diuretics under medical supervision. For Gaisböck syndrome, weight loss, smoking cessation, and blood pressure control matter more than phlebotomy.
Monitoring
Every patient with polycythemia needs regular blood counts, symptom review, and screening for complications. The exact schedule depends on the type and severity.ons are essential to guide treatment adjustments and ensure optimal patient outcomes.
Potential Complications
Polycythemia vera
- Thrombosis (arterial and venous) is the leading cause of morbidity and mortality. Stroke, myocardial infarction, peripheral artery disease, DVT, pulmonary embolism, Budd-Chiari syndrome, and portal vein thrombosis all occur at increased rates.
- Hemorrhage, despite the thickened blood, can affect a minority of patients due to abnormal platelet function.
- Progression to myelofibrosis ("spent phase") affects a subset of patients over time. The marrow scars, blood counts fall, and the spleen enlarges further.
- Transformation to acute myeloid leukemia occurs in a small percentage, with risk slightly raised by older alkylating agents.
- Gout and kidney stones from raised uric acid driven by cell turnover.
- Peptic ulcer disease, possibly linked to histamine release from basophils.
Secondary polycythemia
Complications mix the hyperviscosity risk with whatever the underlying disease brings — worsening hypoxemia, pulmonary hypertension, tumor progression, or cardiac decompensation.
Relative polycythemia
Usually limited to the consequences of the fluid imbalance itself: electrolyte disturbance, acute kidney injury, and (in Gaisböck syndrome) the cardiovascular risks of obesity, smoking, and hypertension
Prognosis
Polycythemia vera prognosis has improved markedly with modern management. With careful hematocrit control, low-dose aspirin, and cytoreductive therapy when indicated, many patients live for decades with good quality of life. Median survival exceeds 15–20 years in well-managed cohorts. Older age at diagnosis, prior thrombosis, and progression to myelofibrosis or leukemia worsen outcomes.
Secondary polycythemia prognosis tracks the underlying disease. Resectable tumors that cause it often see resolution. Chronic lung or heart disease carries its own trajectory, with the polycythemia adding to the risk.
Relative polycythemia typically resolves with correction of the underlying cause and has an excellent outlook overall.
Frequently Asked Questions (FAQs)
What is polycythemia in simple terms?
Polycythemia means too many red blood cells. The blood thickens and flows poorly, raising the risk of clots, strokes, and heart attacks. Causes range from polycythemia vera (a bone marrow disease) to low oxygen states like lung disease or high altitude, to simple dehydration that concentrates the cells.
Is polycythemia life-threatening?
It can be, especially if untreated. Thickened blood (hyperviscosity) raises the risk of dangerous clots in the brain, heart, lungs, and legs. With proper treatment, most patients live long, full lives.
Is polycythemia the same as polycythemia vera?
No. Polycythemia is the finding on a blood test. Polycythemia vera (PV) is one specific cause — a slow-growing blood cancer of the bone marrow driven by a JAK2 mutation. Most polycythemia is not PV.
What is the JAK2 mutation, and why does it matter?
JAK2 is a gene that controls red blood cell production. In PV, more than 95% of patients carry the JAK2 V617F mutation, which keeps the production signal stuck "on." JAK2 testing is one of the most important steps in diagnosing PV.
Why is the hematocrit target less than 45%?
The CYTO-PV trial showed that keeping hematocrit below 45% significantly reduced cardiovascular deaths and major clots in PV [1]. This is now the universal target for both men and women.
What are the latest treatments for polycythemia vera?
Ropeginterferon alfa-2b is now NCCN-preferred first-line cytoreductive therapy [5]. Ruxolitinib is the standard option after hydroxyurea failure, with MAJIC-PV data showing reduced thrombosis [4]. Rusfertide, a hepcidin mimetic, met its endpoints in the phase 3 VERIFY trial in 2025 and is the most anticipated new agent in this space [6].
Can stress cause polycythemia?
Stress can produce a relative polycythemia known as Gaisböck syndrome, where chronic stress, obesity, hypertension, and smoking shrink the plasma volume. Red cell mass is normal; the cells are just more concentrated.
Does drinking water help polycythemia?
For relative polycythemia caused by dehydration, yes — restoring fluid normalizes the counts. For absolute polycythemia, hydration may slightly ease viscosity but does not treat the underlying overproduction. Phlebotomy and cytoreductive therapy remain essential for PV.
Glossary of Related Medical Terms
- Polycythemia (erythrocytosis) — An abnormally high red blood cell count, usually picked up as elevated hemoglobin or hematocrit on a blood test.
- Hematocrit (Hct) — The percentage of blood volume made up of red blood cells.
- Hemoglobin (Hb) — The oxygen-carrying protein inside red blood cells.
- Polycythemia vera (PV) — A bone marrow cancer (myeloproliferative neoplasm) where the marrow makes too many red blood cells, usually due to a JAK2 mutation.
- Myeloproliferative neoplasm (MPN) — A group of blood cancers where the bone marrow overproduces one or more blood cell types.
- JAK2 V617F mutation — A change in the JAK2 gene that switches on red cell production permanently, found in over 95% of PV cases.
- Erythropoietin (EPO) — A hormone made by the kidneys that tells the marrow to produce red blood cells.
- Phlebotomy (venesection) — Removing a unit of blood to lower red cell numbers; the cornerstone of PV treatment.
- Cytoreductive therapy — Medication that lowers blood cell production by the marrow.
- Hydroxyurea (hydroxycarbamide) — An older cytoreductive drug that blocks DNA synthesis in dividing cells.
- Ropeginterferon alfa-2b — A long-acting interferon now preferred as first-line cytoreductive therapy in PV.
- Ruxolitinib — A JAK1/JAK2 inhibitor used when hydroxyurea fails or is not tolerated.
- Rusfertide — An investigational hepcidin mimetic that limits iron availability for red cell production.
- Hyperviscosity —Thickened blood that flows poorly, raising clot risk.
- Aquagenic pruritus — Intense itching after contact with water, common in PV.
- Plethora — A flushed, ruddy complexion from too many red cells in surface vessels.
- Splenomegaly — Enlargement of the spleen.
- Thrombosis — Formation of a blood clot inside a vessel.
- Gaisböck syndrome — Apparent polycythemia from reduced plasma volume, typically in overweight, hypertensive smokers.
- Masked PV — PV in patients whose Hb/Hct sits just below diagnostic thresholds, often missed without bone marrow examination.
Disclaimer: This article is intended for educational and informational purposes only. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Marchioli, R., Finazzi, G., Specchia, G., Cacciola, R., Cavazzina, R., Cilloni, D., De Stefano, V., Elli, E., Iurlo, A., Latagliata, R., Lunghi, F., Lunghi, M., Marfisi, R. M., Musto, P., Masciulli, A., Musolino, C., Cascavilla, N., Quarta, G., Randi, M. L., Rapezzi, D., … CYTO-PV Collaborative Group (2013). Cardiovascular events and intensity of treatment in polycythemia vera. The New England journal of medicine, 368(1), 22–33. https://doi.org/10.1056/NEJMoa1208500
- Arber, D. A., Orazi, A., Hasserjian, R. P., Borowitz, M. J., Calvo, K. R., Kvasnicka, H. M., Wang, S. A., Bagg, A., Barbui, T., Branford, S., Bueso-Ramos, C. E., Cortes, J. E., Dal Cin, P., DiNardo, C. D., Dombret, H., Duncavage, E. J., Ebert, B. L., Estey, E. H., Facchetti, F., Foucar, K., … Tefferi, A. (2022). International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood, 140(11), 1200–1228. https://doi.org/10.1182/blood.2022015850
- Khoury, J. D., Solary, E., Abla, O., Akkari, Y., Alaggio, R., Apperley, J. F., Bejar, R., Berti, E., Busque, L., Chan, J. K. C., Chen, W., Chen, X., Chng, W. J., Choi, J. K., Colmenero, I., Coupland, S. E., Cross, N. C. P., De Jong, D., Elghetany, M. T., Takahashi, E., … Hochhaus, A. (2022). The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia, 36(7), 1703–1719. https://doi.org/10.1038/s41375-022-01613-1
- Harrison, C. N., Nangalia, J., Boucher, R., Jackson, A., Yap, C., O'Sullivan, J., Fox, S., Ailts, I., Dueck, A. C., Geyer, H. L., Mesa, R. A., Dunn, W. G., Nadezhdin, E., Curto-Garcia, N., Green, A., Wilkins, B., Coppell, J., Laurie, J., Garg, M., Ewing, J., … Mead, A. J. (2023). Ruxolitinib Versus Best Available Therapy for Polycythemia Vera Intolerant or Resistant to Hydroxycarbamide in a Randomized Trial. Journal of clinical oncology : official journal of the American Society of Clinical Oncology, 41(19), 3534–3544. https://doi.org/10.1200/JCO.22.01935
- Gisslinger, H., Klade, C., Georgiev, P., Krochmalczyk, D., Gercheva-Kyuchukova, L., Egyed, M., Rossiev, V., Dulicek, P., Illes, A., Pylypenko, H., Sivcheva, L., Mayer, J., Yablokova, V., Krejcy, K., Grohmann-Izay, B., Hasselbalch, H. C., Kralovics, R., Kiladjian, J. J., & PROUD-PV Study Group (2020). Ropeginterferon alfa-2b versus standard therapy for polycythaemia vera (PROUD-PV and CONTINUATION-PV): a randomised, non-inferiority, phase 3 trial and its extension study. The Lancet. Haematology, 7(3), e196–e208. https://doi.org/10.1016/S2352-3026(19)30236-4
- Kremyanskaya, M., Kuykendall, A. T., Pemmaraju, N., Ritchie, E. K., Gotlib, J., Gerds, A., Palmer, J., Pettit, K., Nath, U. K., Yacoub, A., Molina, A., Saks, S. R., Modi, N. B., Valone, F. H., Khanna, S., Gupta, S., Verstovsek, S., Ginzburg, Y. Z., Hoffman, R., & REVIVE Trial Investigators (2024). Rusfertide, a Hepcidin Mimetic, for Control of Erythrocytosis in Polycythemia Vera. The New England journal of medicine, 390(8), 723–735. https://doi.org/10.1056/NEJMoa2308809
- Tefferi, A., & Barbui, T. (2023). Polycythemia vera: 2024 update on diagnosis, risk-stratification, and management. American journal of hematology, 98(9), 1465–1487. https://doi.org/10.1002/ajh.27002
- Barbui, T., Thiele, J., Gisslinger, H., Finazzi, G., Carobbio, A., Rumi, E., Luigia Randi, M., Bertozzi, I., Vannucchi, A. M., Pieri, L., Carrai, V., Gisslinger, B., Müllauer, L., Ruggeri, M., Rambaldi, A., & Tefferi, A. (2014). Masked polycythemia vera (mPV): results of an international study. American journal of hematology, 89(1), 52–54. https://doi.org/10.1002/ajh.23585
- Barbui, T., Tefferi, A., Vannucchi, A. M., Passamonti, F., Silver, R. T., Hoffman, R., Verstovsek, S., Mesa, R., Kiladjian, J. J., Hehlmann, R., Reiter, A., Cervantes, F., Harrison, C., Mc Mullin, M. F., Hasselbalch, H. C., Koschmieder, S., Marchetti, M., Bacigalupo, A., Finazzi, G., Kroeger, N., … Barosi, G. (2018). Philadelphia chromosome-negative classical myeloproliferative neoplasms: revised management recommendations from European LeukemiaNet. Leukemia, 32(5), 1057–1069. https://doi.org/10.1038/s41375-018-0077-1