Key Takeaways
B-cell ALL, or B-cell Acute Lymphoblastic Leukemia, is an aggressive blood cancer in which immature B-lymphocytes (lymphoblasts) multiply uncontrollably in the bone marrow, crowding out healthy blood cells.
- Demographics: It is the most common cancer in children and shows a bimodal age pattern, peaking under age 5 and again after age 65; B-cell ALL accounts for roughly 75–85% of all ALL cases [1,2].
- Symptoms of B-cell ALL ▾: Patients typically present with signs of bone marrow failure (fatigue, infections, bruising) plus extramedullary spread to lymph nodes, liver, spleen, and the central nervous system.
- Diagnosis for B-cell ALL ▾: Diagnosis requires a bone marrow biopsy showing lymphoblasts, plus flow cytometry confirming B-lineage markers (CD19, CD22, CD79a, often CD10 and TdT).
- Treatment of B-cell ALL ▾: Treatment of B-cell ALL has three classic phases (induction, consolidation, and maintenance) and increasingly incorporates immunotherapies such as blinatumomab, inotuzumab ozogamicin, and CD19-directed CAR-T cells [3,4,5]. Pediatric cure rates now exceed 90%; adult 5-year overall survival is approximately 40–50%, with younger adults on pediatric-inspired protocols reaching 60–70% [10].
*Click ▾ for more information
What is Acute Lymphoblastic Leukemia (ALL)?
Acute lymphoblastic leukemia (ALL) is a cancer that starts in the bone marrow, the spongy tissue inside bones where blood cells are made. In ALL, the marrow produces too many immature lymphocytes called lymphoblasts. These blasts cannot fight infection, and they push out the healthy red cells, white cells, and platelets the body needs.
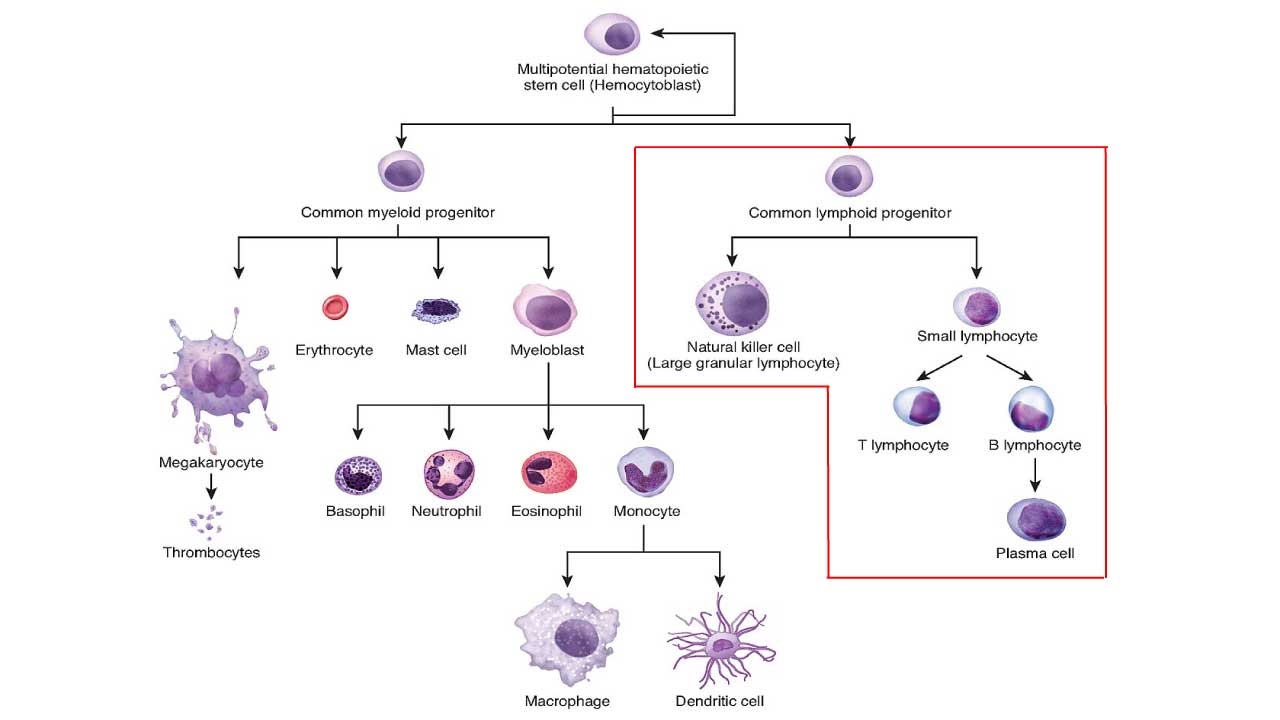
ALL is split into two main types based on which lymphocyte family the cancer comes from: B-cell ALL and T-cell ALL. The split matters because the two behave differently, look different under a microscope, carry different genetic changes, and respond to different drugs.
ALL is the most common cancer of childhood, but it also affects adults, particularly those over 65 [9].
What is B-cell ALL?
B-cell ALL is the more common form of ALL. It accounts for roughly 75–85% of ALL cases across all ages [1]. The disease begins when a single B-cell precursor in the marrow acquires genetic damage that locks it in an immature state. That cell then divides without limit, producing a clone of useless blasts that flood the marrow and spill into the blood.
Most experts now think of B-cell ALL as a two-step process. The first hit is often a chromosomal rearrangement that occurs even before birth (for example, the ETV6-RUNX1 fusion has been found on stored newborn blood spots from children who later developed ALL). The second hit is a postnatal event like additional mutations, possibly triggered by ordinary infections during a key window of immune development that converts the silent pre-leukemic clone into overt disease [6].
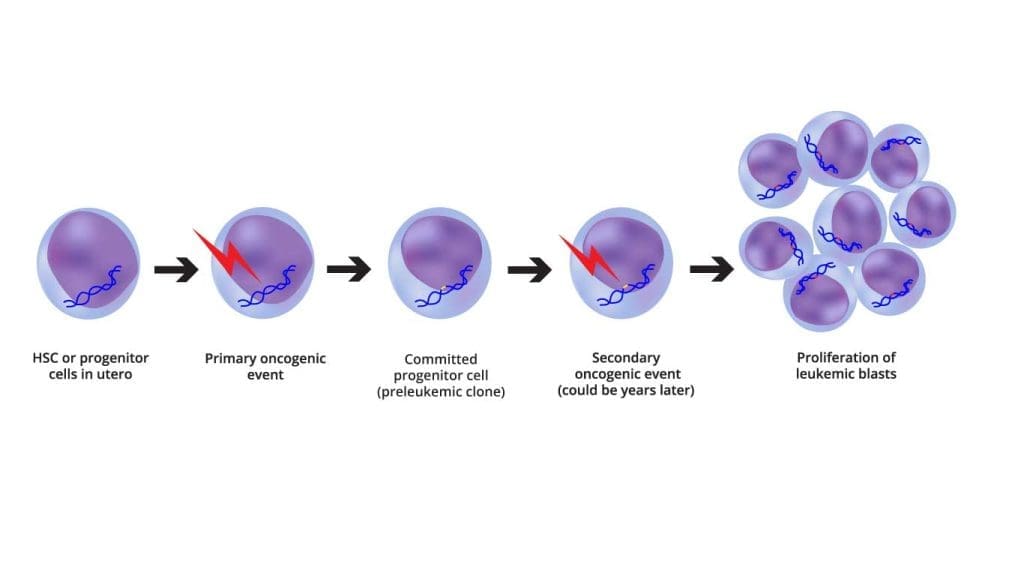
Risk factors and why they matter
The exact cause is unknown in most patients, but several factors increase risk.
- Age. Two peaks: children under 5 and adults over 65. Different genetic landscapes drive each peak.
- Sex. Slight male predominance.
- Down syndrome. Trisomy 21 disrupts B-cell development and is strongly linked to CRLF2 rearrangements and JAK pathway mutations.
- Other genetic syndromes. Klinefelter, Noonan, Li-Fraumeni, neurofibromatosis type 1, and inherited PAX5, ETV6, or IKZF1 mutations all raise risk by destabilizing the genome or DNA repair.
- Ionizing radiation. Including therapeutic and accidental high-dose exposure.
- Benzene and certain pesticides. Linked to acquired marrow damage.
- Family history. A modestly increased risk, but most cases are sporadic.
Clinical Features of B-Cell ALL
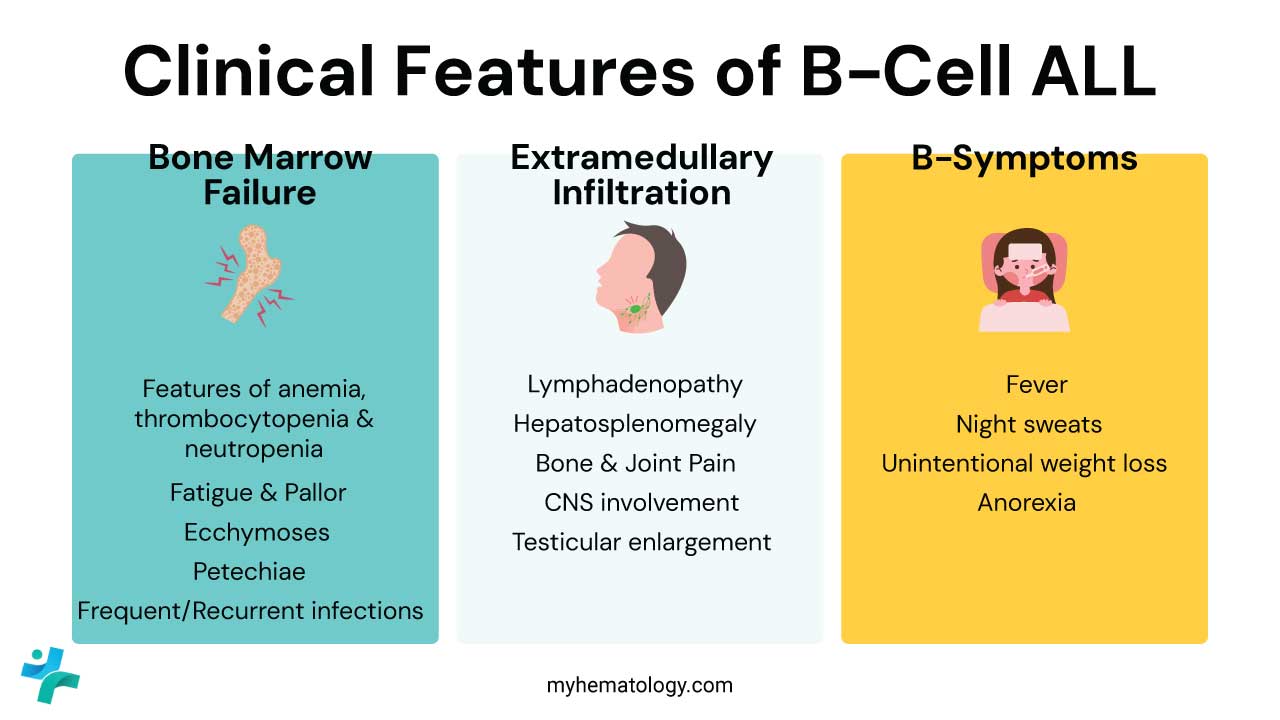
Most symptoms come from two problems: the marrow can no longer make healthy blood cells, and blasts spread to organs outside the marrow.
Bone marrow failure (the "cytopenias")
This is usually how patients first present. As lymphoblasts overrun the marrow, three healthy cell lines collapse:
- Anemia (low red cells) causes fatigue, weakness, breathlessness on exertion, and pale skin and conjunctivae.
- Thrombocytopenia (low platelets) causes easy bruising (ecchymoses), pinpoint red spots (petechiae), bleeding gums, and nosebleeds.
- Neutropenia (low functioning neutrophils) causes frequent or severe infections. Fever in a patient with neutropenia is treated as an emergency until proven otherwise.
Extramedullary infiltration
B-cell ALL likes to settle in lymphoid tissue and so-called sanctuary sites that ordinary chemotherapy struggles to reach.
- Lymphadenopathy. Painless swelling in the neck, armpits, or groin.
- Hepatosplenomegaly. Enlargement of the liver and spleen, which can cause abdominal fullness or a dull ache.
- Bone and joint pain. Caused by marrow expansion and stretching of the periosteum. In children this can look like a limp or refusal to walk, and is sometimes mistaken for growing pains.
- CNS involvement. Headache, vomiting, blurred vision, or cranial nerve palsies (such as a drooping face).
- Testicular enlargement. Painless, firm, usually one-sided. Rare at diagnosis (~2%) but a classic relapse site.
Constitutional "B-symptoms"
These reflect the high metabolic demand of rapidly dividing cancer cells: fever without a clear source, drenching night sweats, unintentional weight loss of more than 10%, and loss of appetite.
| Feature |
Pediatric B-ALL
Typical onset <18 years
|
Adult B-ALL
Typical onset ≥18 years
|
|---|---|---|
| Onset | Often acute (days to weeks) | Can be more insidious |
| Bone Pain | Very common; sometimes mistaken for growing pains | Less prominent |
| WBC Count | Often very high at diagnosis | Variable; pancytopenia frequent |
| Mediastinal Mass | Rare (more typical of T-ALL) | Rare |
| Common Genetics | Favorable ETV6-RUNX1, hyperdiploidy | Higher risk BCR::ABL1, Ph-like, hypodiploidy |
"Red Flag" Clinical Scenarios
- The "limping child." Any child with persistent bone pain and fever should have a complete blood count.
- Superior vena cava syndrome. Facial swelling and respiratory distress from an extremely high blast count or mediastinal mass — an emergency.
- Tumor lysis syndrome at presentation. Decreased urine output, muscle cramps, or arrhythmia from spontaneous breakdown of leukemia cells.
Laboratory Investigations and Diagnosis
The work-up of suspected b-cell ALL moves stepwise from screening blood tests to definitive marrow studies and then to molecular profiling.
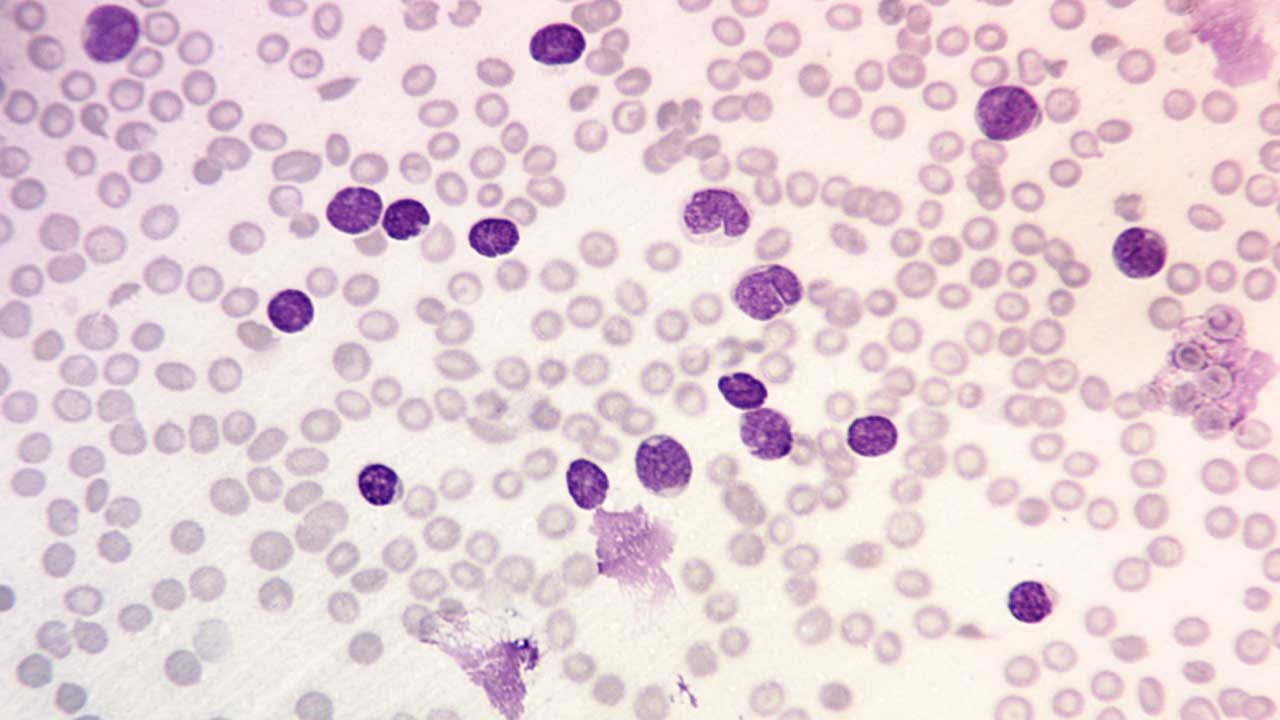
Initial Screening
A complete blood count usually shows pancytopenia, although the white cell count can be low, normal, or extremely high. The peripheral blood smear typically reveals lymphoblasts: small to medium cells with a high nuclear-to-cytoplasmic ratio, fine "dust-like" chromatin, and inconspicuous nucleoli. A "hand-mirror" variant with a tail of cytoplasm is sometimes seen. Smudge cells (fragile blasts that rupture during smearing) are common.
Bone Marrow Examination
A bone marrow aspirate and trephine biopsy are mandatory for definitive diagnosis. The marrow is hypercellular and packed with a uniform population of lymphoblasts.
The traditional 20% blast threshold still applies, but the WHO 2022 classification removes this requirement when defining genetic abnormalities (such as KMT2A rearrangements or BCR::ABL1) are present, because the diagnosis is genetically certain even at lower blast counts [1].
On cytochemistry, B-lymphoblasts are myeloperoxidase (MPO) negative and Sudan Black B negative, but periodic acid–Schiff (PAS) positive, often in a chunky block-like pattern.
Immunophenotyping (Flow Cytometry)
Flow cytometry identifies which family the leukemia belongs to:
- B-lineage markers: CD19, CD22, CD79a (cytoplasmic CD79a is highly specific).
- Immaturity markers: TdT (terminal deoxynucleotidyl transferase), CD34, HLA-DR.
- CD10 (CALLA): Common in childhood B-ALL and a useful prognostic marker.
- Negative for: myeloid markers (MPO, CD13, CD33) and T-cell markers (CD3, CD7).
Cytogenetics and Molecular Diagnostics
Every new B-cell ALL case undergoes karyotyping, FISH, and increasingly next-generation sequencing for risk stratification.
| Genetic Subtype | Significance |
|---|---|
|
t(12;21) [ETV6-RUNX1]
|
Favorable
Most common translocation in children; associated with favorable prognosis.
|
|
High hyperdiploidy (>50 chromosomes)
|
Favorable
Favorable prognosis.
|
|
t(9;22) (BCR::ABL1)
(Ph+)
|
High Risk
High risk; treated with TKIs such as ponatinib.
|
|
KMT2A (MLL) rearranged
|
High Risk
Common in infants; historically very poor, but now targetable by recently FDA-approved menin inhibitors (e.g., revumenib) [11]
|
|
Hypodiploidy (<44 chromosomes)
|
High Risk
High risk.
|
|
Ph-like (BCR::ABL1-like)
|
High Risk
Same gene-expression signature as Ph+ but no t(9;22); often CRLF2 rearrangements or JAK mutations; high risk.
|
Ph-like ALL
Ph-like ALL accounts for around 10–15% of pediatric B-ALL and up to 25% of adolescent and young adult B-ALL, and is enriched in Hispanic populations. Because it carries Ph-like risk without the Philadelphia chromosome, it can only be found by full molecular profiling [7].
Minimal Residual Disease (MRD)
Once treatment starts, "remission" is no longer judged by microscope alone. Measurable (formerly "minimal") residual disease is the most powerful predictor of relapse. Achieving deep, NGS-verified MRD negativity is increasingly allowing patients to safely bypass allogeneic stem cell transplants and their associated toxicities [12]:
- Flow cytometry MRD: The standard rapid test; detects 1 leukemia cell in 10,000.
- NGS-based MRD (e.g., clonoSEQ): The gold standard for critical pivot points; detects 1 in 1,000,000.
A patient who remains MRD-positive after consolidation is a candidate for CAR-T cell therapy or an allogeneic stem cell transplant.
Central Nervous System (CNS) Evaluation
A lumbar puncture at diagnosis examines the cerebrospinal fluid for blasts. CNS involvement is graded CNS-1 (no blasts), CNS-2 (blasts present, low WBC), or CNS-3 (high WBC plus blasts), and influences how aggressively the brain and spinal cord are treated.
Treatment and Management of B-Cell ALL
The goal is complete remission: no detectable leukemia in the body, including by MRD testing. Treatment of B-cell ALL is long, intensive, and tailored to age, fitness, and genetic subtype. There are two broad tracks:
- Philadelphia-negative
- Philadelphia-positive
and a growing role for immunotherapy and CAR-T cells.
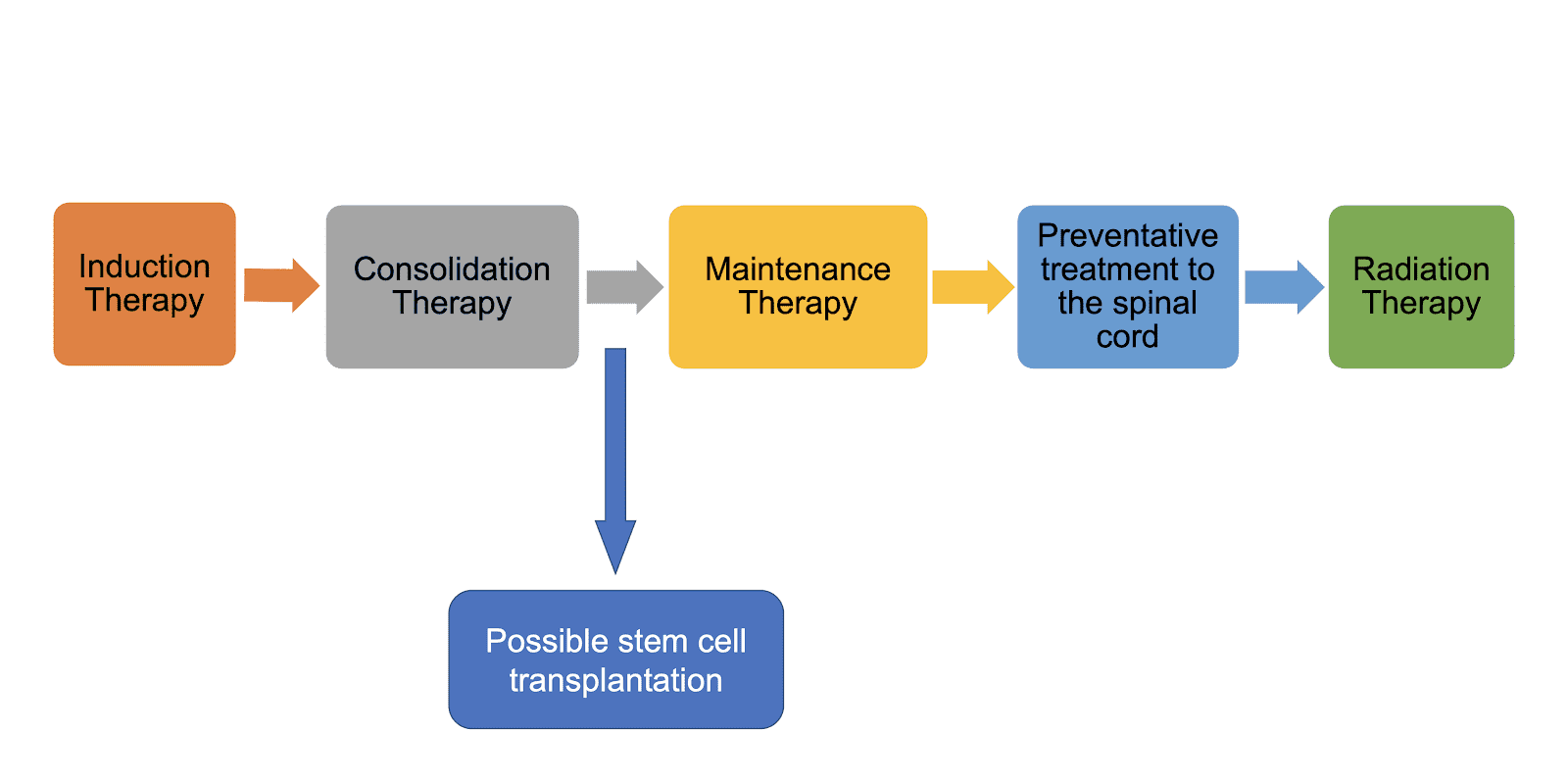
The Classic Three-Phase Protocol (Ph-Negative)
For patients without the Philadelphia chromosome, treatment lasts about 2–3 years.
Phase I — Induction (4 to 6 weeks): Goal: Morphologic Complete Remission. Wipes out visible leukemia (less than 5% blasts in marrow) and restores normal blood counts. The backbone combines vincristine, a corticosteroid, and an anthracycline, with asparaginase in pediatric/adolescent regimens. Rituximab is added for CD20-positive disease [6].
Phase II — Consolidation (Multiple blocks over months): Goal: Eradicate Measurable Residual Disease (MRD). Uses high-dose methotrexate and cytarabine. Based on the landmark ECOG-ACRIN E1910 trial, adding blinatumomab to consolidation is now a proven frontline standard for adult Ph-negative B-ALL that significantly boosts overall survival, even for patients who are already MRD-negative [13].
Phase III — Maintenance (1.5 to 2 years): Goal: Prevent late relapse. Daily oral 6-mercaptopurine and weekly methotrexate. Maintenance is often omitted or shortened if the patient proceeds to CAR-T therapy or a stem cell transplant.
Philadelphia-positive B-cell ALL: the targeted track
The treatment of Ph+ B-ALL has been transformed by tyrosine kinase inhibitors (TKIs). Two recent advances have reshaped practice:
Chemo-free ponatinib + blinatumomab. A phase II trial reported a 3-year overall survival of 88% with this chemo-free combination [3]. The paradigm for Ph+ ALL is rapidly shifting from a promising trial to a preferred frontline option. This proves that highly targeted therapy can yield excellent survival while avoiding the heavy toxicity of traditional cytotoxic chemotherapy, making it especially attractive for older or frail adults.
PhALLCON trial. Frontline ponatinib plus reduced-intensity chemotherapy produced significantly higher MRD-negative complete response rates than imatinib plus the same chemotherapy in newly diagnosed Ph+ ALL. Ponatinib gained accelerated FDA approval for this indication in March 2024 [4].
Immunotherapy & CAR-T cells
Modern immunotherapy has changed what is possible in relapsed and refractory disease, and is steadily moving into frontline treatment.
- Blinatumomab. A bispecific T-cell engager (BiTE) that links the patient's T cells to CD19-positive leukemia cells, forcing an immune attack.
- Inotuzumab ozogamicin. An antibody-drug conjugate that delivers a toxic payload to CD22-positive B cells.
- CAR-T cell therapy. Patient T cells are genetically engineered to recognize CD19 and reinfused.
- Obecabtagene autoleucel (obe-cel; Aucatzyl). FDA-approved on November 8, 2024 for adults with relapsed or refractory B-cell precursor ALL, based on the FELIX trial. Its "fast-off" CD19 binding lowers rates of severe cytokine release syndrome and neurotoxicity compared with earlier products [5].
- Brexucabtagene autoleucel. Earlier-generation CAR-T, used in younger adults and high-risk relapse.
- Tisagenlecleucel. The first CD19 CAR-T approved, used in pediatric and young adult relapsed disease.
CAR-T Timing
While historically reserved for full morphological relapse, CAR-T therapy is increasingly moving earlier in the treatment algorithm. It is now frequently used as a consolidation strategy for high-risk patients who fail to clear MRD upfront, rather than waiting for the disease to fully return.
A practical complication of CD19-directed therapy is antigen escape: leukemia cells that have lost CD19 grow back during or after treatment. CD22-directed and bispecific approaches are being studied to address this.
Side effects
Two immunotherapy toxicities deserve their own definitions:
- Cytokine release syndrome (CRS). A flu-like inflammatory reaction caused by mass T-cell activation, ranging from fever and low blood pressure to life-threatening shock. Treated with the IL-6 blocker tocilizumab and corticosteroids [8].
- Immune effector cell-associated neurotoxicity syndrome (ICANS). Confusion, language difficulty, tremor, or seizures, usually within days of CAR-T infusion. Managed with corticosteroids and supportive care [8].
Supportive Care and Sanctuary Sites
CNS prophylaxis. Because most chemotherapy crosses the blood-brain barrier poorly, intrathecal chemotherapy (methotrexate, cytarabine, or steroids) is given via lumbar puncture throughout treatment. Cranial radiation is now reserved for selected high-risk cases.
Tumor lysis syndrome. Aggressive hydration plus rasburicase (a recombinant urate oxidase) is standard for high-risk patients.
Growth factors. Granulocyte colony-stimulating factor (G-CSF) shortens neutropenia and reduces serious infections.
What Caregivers Can Expect During Treatment
Induction usually means a hospital stay of 4–6 weeks. Most patients have a central line placed for chemotherapy, transfusions, and antibiotics. Hair loss, mouth sores, nausea, and severe fatigue are common. Fever during neutropenia is treated as an emergency, so families learn to take temperatures carefully and to seek care quickly.
Beyond induction, life shifts to outpatient visits, blood counts, and intermittent intrathecal chemotherapy. Fertility preservation is discussed before treatment when possible. Schools, employers, and mental health support all become part of the care plan. Most children and adults reach remission within the first 4–6 weeks of treatment, but the full course extends over years, and emotional support matters as much as the drugs.
Prognosis
Outcomes for B-cell ALL have improved dramatically over the past two decades. Today, more than 90% of children achieve long-term remission. Adult outcomes are improving but remain lower: 5-year overall survival is roughly 40–50% across all ages, and 60–70% in younger adults treated on pediatric-inspired regimens [10].
Prognosis depends on:
- Age. Children do better than adults.
- WBC count at diagnosis. Very high counts predict worse outcomes.
- CNS involvement. Worsens prognosis and requires more intensive CNS-directed therapy.
- Genetics. ETV6-RUNX1 and high hyperdiploidy are favorable; BCR::ABL1, KMT2A rearrangements, hypodiploidy, and Ph-like signatures are unfavorable.
- MRD response. The single strongest predictor of relapse.
B-ALL vs T-ALL
Acute Lymphoblastic Leukemia
Frequently Asked Questions (FAQs)
Is B-cell ALL curable?
Yes, especially in children. Long-term remission rates exceed 90% in pediatric patients with modern protocols [2]. Adult outcomes are improving steadily, with 5-year overall survival around 40–50% overall and 60–70% in younger adults on pediatric-inspired regimens [10].
Is B-cell ALL hereditary?
Most cases are not inherited. They arise from genetic changes acquired during life. A small fraction occur in people with inherited conditions such as Down syndrome or Li-Fraumeni syndrome. Family history can slightly raise risk but rarely causes the disease directly.
What is the difference between B-cell ALL and B-cell lymphoblastic lymphoma?
They are the same disease at different ends of a spectrum. B-cell ALL involves mainly the marrow and blood. B-cell lymphoblastic lymphoma presents as solid masses (usually in lymph nodes or the mediastinum) with less than 25% marrow involvement. Treatment is broadly similar.
Why is B-cell ALL more curable in children than in adults?
Children more often carry favorable genetic subtypes such as ETV6-RUNX1 and high hyperdiploidy, while adults more often have high-risk lesions like the Philadelphia chromosome, Ph-like signatures, and hypodiploidy. Children also tolerate intensive multi-year chemotherapy better.
Can a routine blood test detect B-cell ALL?
A complete blood count can raise suspicion if it shows abnormal white cell counts, anemia, or low platelets. Definitive diagnosis still requires a bone marrow biopsy and flow cytometry to identify B-cell markers.
Why are the CNS and testes called "sanctuary sites"?
Standard chemotherapy crosses the blood-brain barrier and reaches the testes poorly. Leukemia cells can hide there during treatment. Specialized strategies like intrathecal chemotherapy and, when needed, cranial radiation are required to clear them.
Is MRD-negative status the same as being cured?
No. MRD-negative means no leukemia cells are detected by sensitive testing, which is the strongest predictor of long-term remission. Patients still complete maintenance therapy and are monitored for years afterward.
Glossary of Related Medical Terms
- Allogeneic stem cell transplant — A transplant using stem cells from a donor to replace diseased marrow.
- Antigen escape — Leukemia cells lose or hide a marker (such as CD19) that an immunotherapy targets, allowing them to survive treatment.
- Bispecific T-cell engager (BiTE) — An antibody that links T cells to leukemia cells to force an immune attack.
- Blast — An immature, non-functional blood cell.
- Cytokine release syndrome (CRS) — A flu-like inflammatory reaction caused by mass T-cell activation during immunotherapy.
- Cytogenetics — The study of chromosome changes in cancer cells.
- Extramedullary disease — Leukemia found outside the bone marrow.
- Hyperdiploidy — Cancer cells contain more than the normal 46 chromosomes; >50 is favorable.
- ICANS — Brain-related side effects of CAR-T therapy.
- Induction chemotherapy — The first, most intensive phase of treatment.
- Intrathecal chemotherapy — Drugs delivered into the spinal fluid.
- Lymphoblast — An immature lymphocyte.
- Measurable residual disease (MRD) — (Formerly minimal residual disease). The tiny fraction of leukemia cells remaining after treatment, the complete eradication of which is critical for long-term survival.
- Pancytopenia — A drop in red cells, white cells, and platelets together.
- Philadelphia chromosome — The shortened chromosome 22 created by t(9;22), producing BCR::ABL1.
- Ph-like ALL — Behaves like Ph+ ALL but without the t(9;22) translocation.
- Tumor lysis syndrome (TLS) — A metabolic emergency from rapid cancer cell death.
- Tyrosine kinase inhibitor (TKI) — A drug that blocks BCR-ABL1 and similar signaling proteins.
Disclaimer: This article is intended for educational and informational purposes only. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Alaggio, R., Amador, C., Anagnostopoulos, I., Attygalle, A. D., Araujo, I. B. O., Berti, E., Bhagat, G., Borges, A. M., Boyer, D., Calaminici, M., Chadburn, A., Chan, J. K. C., Cheuk, W., Chng, W. J., Choi, J. K., Chuang, S. S., Coupland, S. E., Czader, M., Dave, S. S., de Jong, D., … Xiao, W. (2022). The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia, 36(7), 1720–1748. https://doi.org/10.1038/s41375-022-01620-2
- Inaba, H., & Pui, C. H. (2021). Advances in the Diagnosis and Treatment of Pediatric Acute Lymphoblastic Leukemia. Journal of clinical medicine, 10(9), 1926. https://doi.org/10.3390/jcm10091926
- Jabbour, E., Short, N. J., Jain, N., Huang, X., Montalban-Bravo, G., Banerjee, P., Rezvani, K., Jiang, X., Kim, K. H., Kanagal-Shamanna, R., Khoury, J. D., Patel, K., Kadia, T. M., Daver, N., Chien, K., Alvarado, Y., Garcia-Manero, G., Issa, G. C., Haddad, F. G., Kwari, M., … Kantarjian, H. (2023). Ponatinib and blinatumomab for Philadelphia chromosome-positive acute lymphoblastic leukaemia: a US, single-centre, single-arm, phase 2 trial. The Lancet. Haematology, 10(1), e24–e34. https://doi.org/10.1016/S2352-3026(22)00319-2
- Jabbour, E., Kantarjian, H. M., Aldoss, I., Montesinos, P., Leonard, J. T., Gómez-Almaguer, D., Baer, M. R., Gambacorti-Passerini, C., McCloskey, J., Minami, Y., Papayannidis, C., Rocha, V., Rousselot, P., Vachhani, P., Wang, E. S., Wang, B., Hennessy, M., Vorog, A., Patel, N., Yeh, T., … Ribera, J. M. (2024). Ponatinib vs Imatinib in Frontline Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia: A Randomized Clinical Trial. JAMA, 331(21), 1814–1823. https://doi.org/10.1001/jama.2024.4783
- Roddie, C., Sandhu, K. S., Tholouli, E., Logan, A. C., Shaughnessy, P., Barba, P., Ghobadi, A., Guerreiro, M., Yallop, D., Abedi, M., Pantin, J. M., Yared, J. A., Beitinjaneh, A. M., Chaganti, S., Hodby, K., Menne, T., Arellano, M. L., Malladi, R., Shah, B. D., Mountjoy, L., … Jabbour, E. (2024). Obecabtagene Autoleucel in Adults with B-Cell Acute Lymphoblastic Leukemia. The New England journal of medicine, 391(23), 2219–2230. https://doi.org/10.1056/NEJMoa2406526
- Brown, P. A., Shah, B., Advani, A., Aoun, P., Boyer, M. W., Burke, P. W., DeAngelo, D. J., Dinner, S., Fathi, A. T., Gauthier, J., Jain, N., Kirby, S., Liedtke, M., Litzow, M., Logan, A., Luger, S., Maness, L. J., Massaro, S., Mattison, R. J., May, W., … Campbell, M. (2021). Acute Lymphoblastic Leukemia, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. Journal of the National Comprehensive Cancer Network : JNCCN, 19(9), 1079–1109. https://doi.org/10.6004/jnccn.2021.0042
- Jain, N., Roberts, K. G., Jabbour, E., Patel, K., Eterovic, A. K., Chen, K., Zweidler-McKay, P., Lu, X., Fawcett, G., Wang, S. A., Konoplev, S., Harvey, R. C., Chen, I. M., Payne-Turner, D., Valentine, M., Thomas, D., Garcia-Manero, G., Ravandi, F., Cortes, J., Kornblau, S., … Konopleva, M. (2017). Ph-like acute lymphoblastic leukemia: a high-risk subtype in adults. Blood, 129(5), 572–581. https://doi.org/10.1182/blood-2016-07-726588
- Lee, D. W., Santomasso, B. D., Locke, F. L., Ghobadi, A., Turtle, C. J., Brudno, J. N., Maus, M. V., Park, J. H., Mead, E., Pavletic, S., Go, W. Y., Eldjerou, L., Gardner, R. A., Frey, N., Curran, K. J., Peggs, K., Pasquini, M., DiPersio, J. F., van den Brink, M. R. M., Komanduri, K. V., … Neelapu, S. S. (2019). ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation, 25(4), 625–638. https://doi.org/10.1016/j.bbmt.2018.12.758
- Siegel, D. A., Henley, S. J., Li, J., Pollack, L. A., Van Dyne, E. A., & White, A. (2017). Rates and Trends of Pediatric Acute Lymphoblastic Leukemia - United States, 2001-2014. MMWR. Morbidity and mortality weekly report, 66(36), 950–954. https://doi.org/10.15585/mmwr.mm6636a3
- Sasaki, K., Jabbour, E., Short, N. J., Jain, N., Ravandi, F., Pui, C. H., & Kantarjian, H. (2021). Acute lymphoblastic leukemia: A population-based study of outcome in the United States based on the surveillance, epidemiology, and end results (SEER) database, 1980-2017. American journal of hematology, 96(6), 650–658. https://doi.org/10.1002/ajh.26156
- Lynch, E. J., Faro, S. J. E., Lindstrom, A. M., Sethi, N. A., Wang, W. Y., & Seligson, N. D. (2025). Revumenib for Relapsed or Refractory Acute Leukemia With a KMT2A Translocation. The Annals of pharmacotherapy, 59(12), 1108–1118. https://doi.org/10.1177/10600280251341279
- Saygin, C., Cannova, J., Stock, W., & Muffly, L. (2022). Measurable residual disease in acute lymphoblastic leukemia: methods and clinical context in adult patients. Haematologica, 107(12), 2783–2793. https://doi.org/10.3324/haematol.2022.280638
- Litzow, M. R., Sun, Z., Mattison, R. J., Paietta, E. M., Roberts, K. G., Zhang, Y., Racevskis, J., Lazarus, H. M., Rowe, J. M., Arber, D. A., Wieduwilt, M. J., Liedtke, M., Bergeron, J., Wood, B. L., Zhao, Y., Wu, G., Chang, T. C., Zhang, W., Pratz, K. W., Dinner, S. N., … Tallman, M. S. (2024). Blinatumomab for MRD-Negative Acute Lymphoblastic Leukemia in Adults. The New England journal of medicine, 391(4), 320–333. https://doi.org/10.1056/NEJMoa2312948