TL;DR
The coagulation cascade is a complex series of biochemical reactions that occur in the blood when a vessel is injured. It’s a tightly regulated process that involves numerous proteins, called coagulation factors, working together to form a blood clot and stop bleeding.
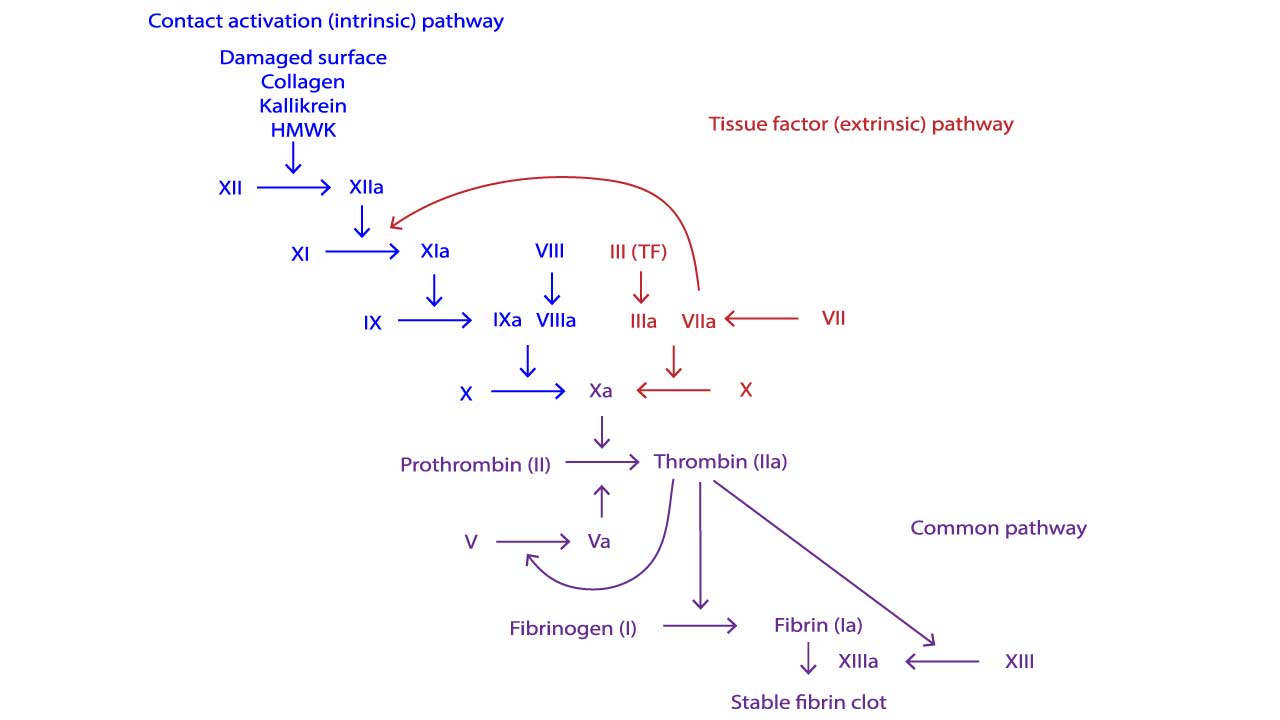
- Two Pathways ▾: The intrinsic and extrinsic pathways, triggered by internal and external damage respectively, converge in the common pathway, culminating in fibrin clot formation.
- Key players ▾: Zymogens, serine proteases, cofactors, calcium ions.
- Fibrin Formation ▾: The fibrin clot acts as a natural plug, stopping blood loss and allowing wound healing to begin.
- Regulation and Inhibition ▾: The process is tightly controlled by natural anticoagulants like antithrombin III, preventing excessive clot formation and maintaining balance.
- Related Bleeding Disorders: Hemophilia, factor V deficiency, protein C/S deficiency etc.
*Click ▾ for more information
Introduction
Hemostasis is the body’s remarkable process of stopping blood flow at a site of injury. It’s a delicate balance between maintaining smooth blood flow throughout the body and preventing excessive bleeding when a vessel is damaged. This article covers the importance of secondary hemostasis or the coagulation cascade in more detail.
The Two Phases of Hemostasis
Hemostasis operates in two distinct phases, working together seamlessly to seal the breach.
- Primary Hemostasis: This is the initial response, activated within seconds of injury. Platelets stick to the damaged vessel wall like sticky patches, forming a temporary platelet plug to slow the blood flow.
- Secondary Hemostasis (Coagulation Cascade): This is the reinforcement phase, taking over from platelets within a few minutes. A complex cascade of proteins called coagulation factors kicks in, creating a fibrin mesh that strengthens and stabilizes the platelet plug into a permanent clot.
The blood coagulation cascade plays a vital role in maintaining blood flow and preventing excessive bleeding. It’s like a powerful backup system, ensuring the platelet plug is adequately reinforced and stabilized to effectively seal the wound.
Hemostasis is a finely balanced process. While clotting is crucial to stop bleeding, excessive clotting can be dangerous, blocking blood flow and causing tissue damage. To prevent this, the body has a built-in system of anticoagulants, proteins that work to break down and remove clots once they are no longer needed. It’s like a constant tug-of-war, ensuring the clot forms where and when needed but doesn’t overstay its welcome.
Coagulation Factors in the Coagulation Cascade
- Zymogens: These are inactive precursors of coagulation factors. They wait patiently in the blood until a trigger, like an injury, sets them off.
- Serine proteases: These are enzymes that snip specific protein bonds in other factors, activating them.
- Cofactors: These are proteins that bind to and stabilize serine proteases, ensuring they work efficiently.
- Calcium ions: These are the “spark plugs,” essential minerals that bind to specific sites on coagulation factors, enabling them to function properly.
Numbering system and nomenclature used for coagulation factors
Coagulation factors in the coagulation cascade might seem like a jumble of names, but there’s actually a logic behind it! They’re numbered based on their order of discovery, with Roman numerals for historical reasons (e.g., factor VII was discovered before factor VIII). Sometimes, lowercase letters are added to indicate different activation states (e.g., factor Xa is the activated form of factor X).
Main coagulation factors, their activation mechanisms, and functions.
| Factor | Activation Mechanism | Function |
| Tissue Factor (TF) | Injury exposure | Initiates extrinsic pathway |
| Factor VII (FVII) | TF binding, calcium | Activates factor X |
| Factor X (FX) | FVIIa cleavage, calcium | Activates factor IX and prothrombin |
| Factor IX (FIX) | FXa cleavage, calcium, factor VIIIa | Activates factor X |
| Factor VIII (FVIII) | Factor IXa cleavage, calcium | Cofactor for factor IXa |
| Prothrombin (FII) | FXa cleavage, calcium | Converted to thrombin |
| Thrombin (FIIa) | Cleaves fibrinogen | Forms fibrin clot, activates factor XIII |
| Fibrinogen (FI) | Thrombin cleavage | Forms fibrin mesh, strengthens clot |
| Factor XIII (FXIII) | Thrombin activation, calcium | Stabilizes fibrin clot by cross-linking |
| Protein C (PC) | Thrombin activation | Inhibits factors Va and VIIIa, prevents excessive clotting |
Coagulation Cascade
Intrinsic Pathway
The intrinsic pathway of the coagulation cascade is activated by internal factors or contact with collagen following injury. Once triggered, factor XII activates factor XI, which then activates factor IX. Factor IX, along with factor VIII and calcium, forms a complex that activates factor X.
This activated factor X, alongside factor Va and calcium, then converts prothrombin (factor II) into thrombin, the enzyme that drives clot formation by converting fibrinogen into fibrin, the mesh of fibers that forms the clot.
Extrinsic Pathway
The extrinsic pathway of the coagulation cascade is triggered by external injury, initiates clotting faster than the intrinsic pathway. When tissue factor (factor III) exposed by damage comes in contact with circulating factor VII, they activate factor X. This activated factor X then joins forces with factors Va and calcium to convert prothrombin (factor II) into thrombin, the enzyme responsible for clot formation. This kickstarts the common pathway of the coagulation pathway, leading to fibrin clot formation.
Common Pathway
In a so-called prothrombinase complex, activated factor X together with activated factor V promotes a downstream enzymatic cleavage of prothrombin in the coagulation cascade, which yields small amounts of thrombin.
Thrombin is a pleiotropic, central coagulation enzyme that not only converts fibrinogen into fibrin but also has a substantial role in the activation of platelets and activates factor XIII to induce fibrin polymerization, a fundamental process for the formation of a stable clot, or thrombus.
By supporting positive-feedback activation of the upstream factors V, VIII, and XI, thrombin plays a crucial part in the amplification and propagation phases of coagulation .
The activated platelet surface is also a critical catalyst for the coagulation cascade.
Platelets actively participate in the clotting process by introducing extra amounts of tissue factor, factor V, fibrinogen, and factor XIII into the system, derived from various local sources (fibrinogen and factors V and XIII stored in α granules), and facilitating the direct activation of factor XI by thrombin and the subsequent activation of factor IX on the platelet surface.
Factor IXa forms the so-called tenase complex together with factor VIIIa, thereby igniting a burst of additional thrombin generation, which is essential in forming sufficient fibrin and sealing the defect.
Fibrin Formation and Clot Stabilization
The climax of hemostasis lies in the transformation of a soluble protein called fibrinogen into a robust fibrin mesh that seals the wound. This intricate process involves two crucial players: activated thrombin and activated factor XIII, all meticulously orchestrated by calcium ions.
From Fibrinogen to Fibrin Monomers
Thrombin cleaves fibrinogen, a large protein in the plasma, into smaller units called fibrin monomers. This cleavage isn’t haphazard. Thrombin targets specific peptide bonds within fibrinogen, releasing fibrinopeptides A and B. These peptides act like anchors, keeping the monomers soluble and preventing premature polymerization.
Thrombin also exposes hidden binding sites on the fibrin monomers. These sites, like molecular velcro, allow the monomers to connect with each other, setting the stage for the next critical step in the coagulation cascade.
Building a Stable Mesh
Activated factor XIII, another vital player, binds to exposed lysine residues on the fibrin monomers, acting like a molecular sewing kit. Factor XIII then catalyzes the formation of covalent bonds between these lysines, stitching the fibrin monomers together into a stable, insoluble mesh. This fibrin mesh is the core of the clot, the physical barrier that plugs the leak and prevents further blood loss.
Calcium Ions
Calcium ions, though not directly involved in the cleavages or cross-linking, play a crucial supporting role. They bind to specific sites on both thrombin and factor XIII, stabilizing their structures and facilitating their interactions with fibrinogen and fibrin monomers.
The conversion of fibrinogen to fibrin and its subsequent cross-linking are crucial for several reasons:
- Clot strength and stability: The fibrin mesh provides a strong, insoluble barrier against blood flow, ensuring the clot remains intact until the wound heals.
- Localized clot formation: The process is tightly regulated by thrombin and factor XIII, preventing unwanted clot formation in the vasculature.
- Wound healing: The fibrin mesh creates a scaffold for tissue repair, promoting cell migration and blood vessel formation at the wound site.
Cell-based Model
The traditional waterfall model of the coagulation cascade, with its distinct intrinsic and extrinsic pathways, has served us well for decades. However, recent advancements have led to the development of a more nuanced and physiologically accurate model: the cell-based model.
This revised model of the coagulation cascade emphasizes the crucial role of cells in orchestrating the coagulation process, offering a more comprehensive understanding of hemostasis in vivo.
The Traditional Model
- Independent Pathways: Imagine two separate waterfalls, intrinsic and extrinsic, converging to form a large clot. The intrinsic pathway starts within the vessel, while the extrinsic pathway is triggered by tissue damage.
- Plasma-Centric: Factors dance primarily in the plasma, with minimal interaction with cells. They activate each other in a domino-like fashion, ultimately leading to fibrin clot formation.
- Emphasis on Factors: Each step revolves around specific coagulation factors, numbered and neatly categorized (e.g., factor VIII, factor XIII).
Key Features of the Cell-Based Model
- Cellular Control: The initiation and amplification of coagulation are primarily localized on the surfaces of activated platelets and vascular endothelial cells, rather than occurring freely in the plasma.
- Three Overlapping Phases: The coagulation cascade is divided into three phases: initiation, amplification, and propagation, instead of the traditional two pathways. Each phase involves specific cell types and interactions.
- Tissue Factor (TF): This key initiator is not just present in injured tissues but also expressed by activated endothelial cells and circulating monocytes, highlighting its broader role in triggering coagulation.
- Fibrinogen Binding: Platelets play a crucial role in confining the fibrin clot formation to the site of injury by binding fibrinogen to their surfaces.
Differences from the Traditional Model
- No Distinct Pathways: The cell-based model emphasizes the interconnectedness and overlap between the traditional intrinsic and extrinsic pathways. Both pathways contribute to the same phases, occurring on the surfaces of activated cells.
- Emphasis on Platelets: Platelets are not just passive participants but actively contribute to the initiation, amplification, and propagation of the cascade through their surface-based reactions.
- Localized Control: Coagulation is tightly localized to the site of injury, minimizing the risk of inappropriate clot formation elsewhere in the vasculature.
Benefits of the Cell-Based Model
- Improved Understanding of Hemostasis: The cell-based model provides a more realistic representation of how coagulation occurs in vivo, leading to a better understanding of physiological hemostasis.
- Diagnosis and Treatment of Bleeding Disorders: This model can offer new insights into the pathogenesis of bleeding disorders and aid in the development of targeted therapies.
- Personalized Medicine: By understanding the cellular and molecular mechanisms involved in blood coagulation, we can pave the way for personalized medicine approaches to bleeding and thrombotic disorders.
The cell-based model doesn’t completely discard the traditional waterfall framework of the coagulation cascade. They represent different viewpoints: waterfall emphasizes the sequence of reactions, while cell-based focuses on the actors and the stage.
Cell-based Model of Coagulation
The cell-based model of coagulation is the modern, physiologically accurate view of how blood clotting occurs within the body, as it accounts for the critical role of cell surfaces. This model replaces the traditional cascade hypothesis and outlines the process in three overlapping stages: initiation, amplification, and propagation.
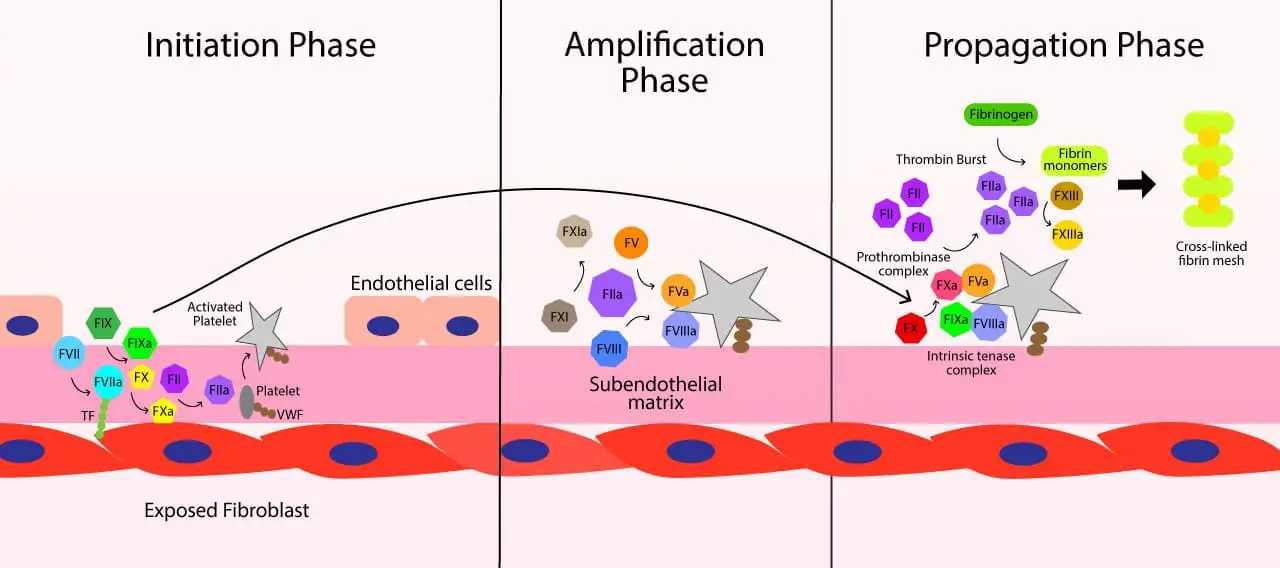
1. Initiation Phase
This phase begins immediately after a blood vessel is injured. The process is started on the surface of cells that express tissue factor (TF), such as fibroblasts in the subendothelial layer of the vessel wall.
- When the vessel is damaged, tissue factor is exposed to the bloodstream.
- Circulating factor VII (FVII) binds to this exposed tissue factor and is converted to its active form, factor VIIa (FVIIa).
- The resulting TF-FVIIa complex activates a small amount of factor X (FX) to factor Xa (FXa), and a small amount of factor IX (FIX) to factor IXa (FIXa).
- The small amount of FXa then binds to the surface of the tissue factor-bearing cell and converts a trace amount of prothrombin (factor II) into thrombin (FIIa). This initial amount of thrombin is too small to form a stable clot, but it is essential to kickstart the next phase.
2. Amplification Phase
The purpose of this stage is to prepare the platelets and cofactors for the large-scale production of thrombin. It is triggered by the small amount of thrombin generated during the initiation phase.
- The trace amount of thrombin activates platelets, causing them to change shape and expose a negatively charged, phospholipid-rich surface (specifically, phosphatidylserine). This activated platelet surface will serve as the platform for the reactions in the final stage.
- This thrombin also activates cofactors, factor V (FV) and factor VIII (FVIII), which then localize on the activated platelet surface.
- Furthermore, the thrombin activates factor XI (FXI) to factor XIa (FXIa), which will later play a role in amplifying the process.
3. Propagation Phase
This is the stage where the majority of thrombin is generated, leading to the formation of a stable blood clot. This happens on the surface of the activated platelets from the amplification phase.
- On the platelet surface, factor IXa (from the initiation phase) binds to factor VIIIa (activated during amplification) to form the intrinsic tenase complex.
- This complex then efficiently activates a large amount of factor X to factor Xa (FXa).
- The newly formed FXa, along with its cofactor factor Va (activated during amplification), forms the prothrombinase complex on the platelet surface.
- The prothrombinase complex then converts a massive amount of prothrombin into thrombin, creating a “burst” of thrombin generation that is crucial for forming the clot.
- The final thrombin burst then cleaves fibrinogen into insoluble fibrin monomers, and activates factor XIII (FXIII). Factor XIII then cross-links these fibrin monomers, forming a stable fibrin mesh that holds the platelet plug in place and creates a durable blood clot.
Comparison of The Traditional Coagulation Cascade vs Cell-Based Models
| Feature/Aspect | Traditional Coagulation Cascade Model | Cell-Based Model |
| Initiation | Factor XII (intrinsic) or Tissue Factor (extrinsic) | Tissue Factor (TF)-bearing cells exposed at injury site |
| Amplification | Sequential activation of factors in fluid phase | Occurs on activated platelet surfaces, localizing reactions |
| Role of Cells | Limited explicit role; focus on plasma proteins | Central; TF-bearing cells initiate, platelets amplify, endothelial cells regulate |
| Physiological Relevance | Describes in vitro laboratory tests; less accurate in vivo | Describes in vivo coagulation more accurately |
| Key Factors Emphasized | All factors in pathways equally critical; Factor XII initiates intrinsic | Factor XII less critical in vivo; Factor VIIa/TF complex is major initiator |
| Clinical Test Correlation | PT (extrinsic), aPTT (intrinsic) directly reflect pathways | Tests reflect plasma factor levels, not full in vivo process |
Coagulation Inhibition
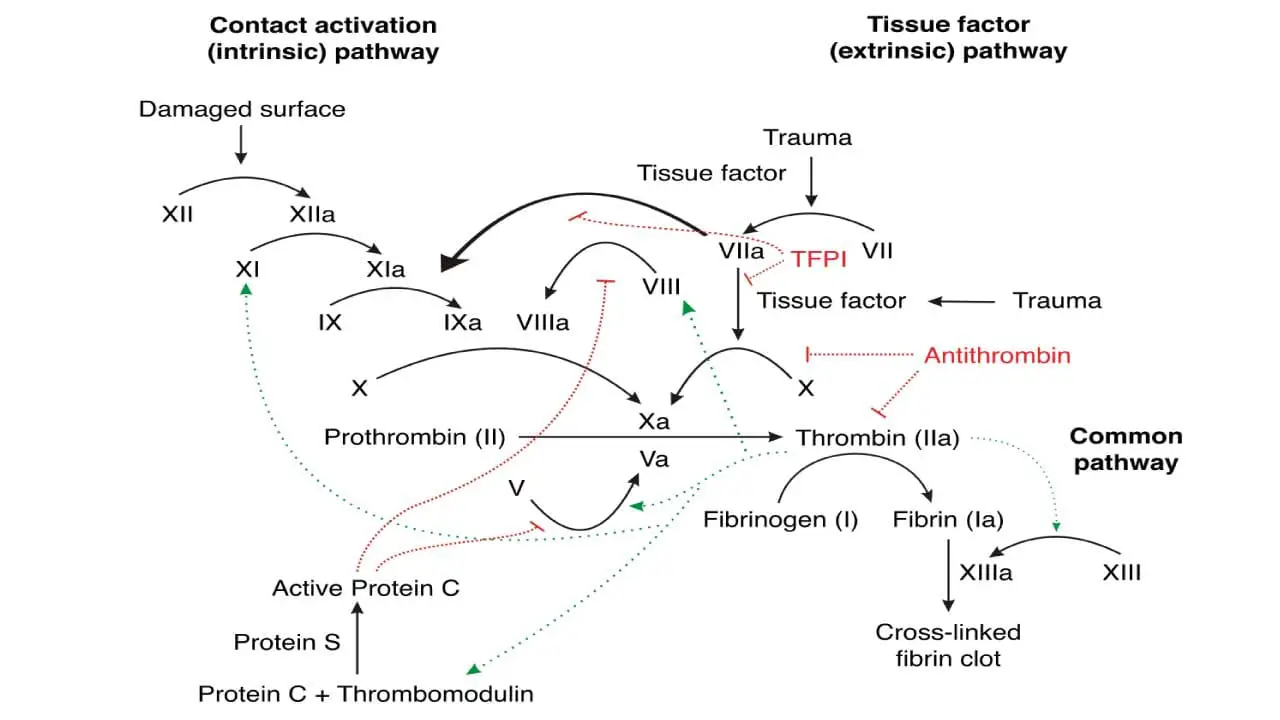
It is just as important that coagulation be limited to the site of injury, because its continued extension to adjacent normal vessels could occlude blood flow which may be fatal. Thus there are several molecules and pathways which limit the extension of clot formation.
- Tissue factor pathway inhibitor (TFPI) is synthesized in endothelial cells and is present in plasma and platelets. It blocks the extrinsic pathway after its initial activation by binding first to factor Xa. This binary factor Xa/TFPI complex then docks onto and inactivates factor VIIa/tissue factor complexes, shutting down the extrinsic pathway of the coagulation cascade after a small amount of factor Xa and subsequently thrombin, have been generated. This is sufficient to activate factor VIII and factor C, enabling the intrinsic pathway to provide a burst of procoagulant activity.
- Antithrombin is the most important plasma protease inhibitor in regulating hemostasis. Upon binding to endogenous glycoconjugates on the surface of endothelial cells or the anticoagulant heparin, antithrombin undergoes a change in conformation that vastly increases its activity as a serine protease inhibitor. It inactivates not only thrombin but also factors VIIa, IXa, Xa and XIa as they are carried in the blood away from the developing thrombus.
- Protein C, in the presence of its cofactor, protein S, is activated by thrombin bound to thrombomodulin on the surface of normal endothelial cells. Protein C and protein S both undergo vitamin K-dependent carboxylation and share other structural features with factors VII, IX, X and prothrombin. Activated protein C reduces the coagulation cascade by specifically inactivating factors Va and VIIIa.
- Thrombomodulin is present on the surface of vascular cells. Thrombomodulin binds to thrombin with high affinity, forming a complex that effectively neutralizes its clotting activity. Thrombomodulin-thrombin complex activates protein C, a potent anti-coagulation agent. Protein C then inactivates factors V and VIII, key players in the coagulation cascade, further dampening clot formation. Beyond stopping clot formation, thrombomodulin plays a role in breaking down existing clots. It activates thrombin-activatable fibrinolysis inhibitor (TAFI), an enzyme that enhances the activity of plasmin, the main player in fibrinolysis.
Dysfunction of the Coagulation Cascade
The coagulation cascade is a finely tuned series of enzymatic reactions essential for forming a stable blood clot (secondary hemostasis) to prevent excessive bleeding. When this intricate system malfunctions, it can lead to two primary and opposing clinical outcomes: bleeding disorders (hypocoagulability), where clotting is insufficient, or thrombotic disorders (hypercoagulability), where clotting is excessive.
Bleeding Disorders (Hypocoagulability)
Bleeding disorders arise when there is an inadequate formation of fibrin, the stable meshwork that reinforces the initial platelet plug. These can be inherited or acquired.
Inherited Coagulation Factor Deficiencies
Deficiencies in specific coagulation factors can severely impair the coagulation cascade, leading to prolonged bleeding episodes.
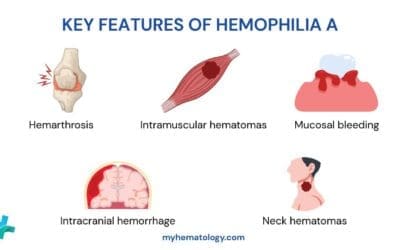
- Hemophilia A: Caused by a deficiency in Factor VIII, which is crucial for the intrinsic pathway of the coagulation cascade.
- Hemophilia B: Results from a deficiency in Factor IX, also vital for the intrinsic pathway.
- Factor VII Deficiency: Affects the extrinsic pathway of the coagulation cascade.
- Factor X Deficiency: Impacts the common pathway, as Factor X is the point where intrinsic and extrinsic pathways converge in the coagulation cascade.
- Factor XIII Deficiency: Leads to unstable clots because Factor XIII is responsible for cross-linking and stabilizing fibrin strands.
- Congenital Afibrinogenemia: A rare condition characterized by a lack of Factor I (fibrinogen), the precursor to fibrin.
- While Factor XII is part of the intrinsic pathway, its deficiency typically does not lead to bleeding, as the coagulation cascade can be reinforced by thrombin’s positive feedback on other factors.
Acquired Coagulation Factor Deficiencies
These conditions develop over time due to other underlying diseases or nutritional deficiencies.
- Chronic Liver Disease: The liver is the primary site of production for many coagulation factors (e.g., Factor I, II, V, VII, IX, X, XI, XII, XIII). Impaired liver function can lead to a depletion of these factors, resulting in defective coagulation.
- Vitamin K Deficiency: Vitamin K is essential for the synthesis of several key coagulation factors, including Factors II (prothrombin), VII, IX, and X, as well as Protein C and Protein S. Deficiency can lead to impaired clotting, with Factor VII being particularly sensitive due to its short half-life.
- Disseminated Intravascular Coagulation (DIC): A complex, life-threatening condition where the coagulation cascade is excessively activated throughout the body, leading to widespread microclot formation. This consumes coagulation factors and platelets, paradoxically resulting in severe bleeding.
Diagnostic Assessment for Bleeding Disorders
Laboratory tests are crucial for identifying specific pathway deficiencies.
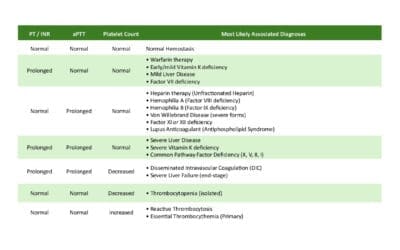
- Prothrombin Time (PT): Measures the time taken for fibrin to form via the extrinsic and common pathways of the coagulation cascade. It assesses Factors VII, X, V, prothrombin (Factor II), and fibrinogen (Factor I). A prolonged PT can indicate deficiencies in these factors, liver disease, or Vitamin K deficiency.
- Activated Partial Thromboplastin Time (APTT): Measures the time taken for fibrin to form via the intrinsic and common pathways of the coagulation cascade. It assesses Factors XII, XI, IX, VIII, X, V, prothrombin, and fibrinogen. A prolonged APTT suggests deficiencies in these factors, such as in hemophilia A or B.
Thrombotic Disorders (Hypercoagulability)
Thrombotic disorders, or hypercoagulability, occur when there is an excessive or inappropriate formation of blood clots (thrombosis). This can be due to overactivity of procoagulant factors or deficiencies in natural anticoagulant mechanisms.
Inherited Thrombophilias
These are genetic predispositions to excessive clotting.
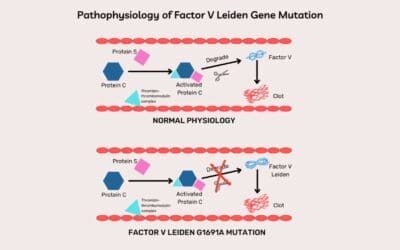
- Factor V Leiden: A common genetic mutation in Factor V that makes it resistant to inactivation by Protein C, a natural anticoagulant. This leads to prolonged activity of Factor Va, promoting excessive thrombin generation and increased clotting risk.
- Prothrombin Thrombophilia (Prothrombin G20210A): A genetic mutation that leads to increased levels of prothrombin (Factor II), resulting in higher thrombin generation and an elevated risk of thrombosis.
Deficiencies in Natural Anticoagulants
The body has built-in mechanisms to regulate and limit clot formation. Deficiencies in these inhibitors can lead to uncontrolled clotting.
- Antithrombin (AT): A natural anticoagulant produced by the liver that primarily inactivates thrombin (Factor IIa) and Factor Xa, as well as Factors XIa and XIIa. Deficiency leads to increased clotting risk.
- Protein C and Protein S: These are vitamin K-dependent proteins that work together to inactivate Factors Va and VIIIa, thereby dampening the coagulation cascade. Deficiencies can result in thrombotic tendencies.
- Tissue Factor Pathway Inhibitor (TFPI): The main inhibitor of the extrinsic pathway, TFPI inactivates the Factor VIIa-Tissue Factor complex and Factor Xa. A deficiency could lead to uncontrolled initiation of coagulation.
Understanding the dysfunction of the coagulation cascade is critical for diagnosing and managing a wide range of bleeding and clotting disorders, ensuring appropriate treatment strategies to maintain hemostatic balance.
Coagulation Cascade Evaluation
Evaluating the coagulation cascade in a laboratory setting is crucial for diagnosing bleeding or clotting disorders, monitoring anticoagulant therapies, and assessing a patient’s hemostatic status, particularly before surgical procedures. These tests measure various proteins and their functions within the complex clotting system.
Screening Coagulation Tests
These are the most commonly performed tests to assess the overall function of the coagulation pathways.
Prothrombin Time (PT) and International Normalized Ratio (INR)
The PT assesses the extrinsic and common pathways of the coagulation cascade. It measures the time it takes for fibrin to form after adding calcium and thromboplastin to a plasma sample. The factors evaluated include Factor VII, Factor X, Factor V, Factor II (prothrombin), and Factor I (fibrinogen).
- Normal Range: Typically 9-12 seconds or 11-15 seconds. The INR is a standardized correction of the PT to account for variations in reagents between laboratories, allowing for international comparison of results. A normal INR is usually between 0.9 and 1.3.
A prolonged PT/INR can indicate deficiencies in the factors it measures, such as Factor VII deficiency, Vitamin K deficiency (as Factor VII has the shortest half-life), or chronic liver disease, which impairs the synthesis of many clotting factors. It is also used to monitor the effectiveness of warfarin therapy, an anticoagulant that inhibits Vitamin K-dependent factors.
Activated Partial Thromboplastin Time (APTT)
The APTT evaluates the intrinsic and common pathways of the coagulation cascade. It assesses factors including Factor XII, Factor XI, Factor IX, Factor VIII, Factor X, Factor V, Factor II (prothrombin), and Factor I (fibrinogen).
- Normal Range: Typically 23-38 seconds or 25-40 seconds.
A prolonged APTT can suggest deficiencies in factors of the intrinsic pathway, such as Hemophilia A (Factor VIII deficiency) or Hemophilia B (Factor IX deficiency). It can also be prolonged in conditions like von Willebrand disease, disseminated intravascular coagulation (DIC), or the presence of inhibitors like lupus anticoagulant. The APTT is primarily used to monitor unfractionated heparin therapy.
Thrombin Time (TT)
The TT directly measures the final step of the coagulation cascade: the conversion of fibrinogen (Factor I) into fibrin by thrombin (Factor IIa). It is not routinely performed as part of a standard coagulation screen.
- Normal Range: Approximately 12 to 19 seconds or less than 24 seconds.
A prolonged TT can indicate low, high, or dysfunctional fibrinogen levels. It can also be prolonged by the presence of certain medications like heparin or argatroban, liver diseases, some cancers (e.g., multiple myeloma), or conditions like DIC. A reptilase time test can be performed alongside TT to differentiate if the prolongation is due to heparin or a fibrinogen disorder.
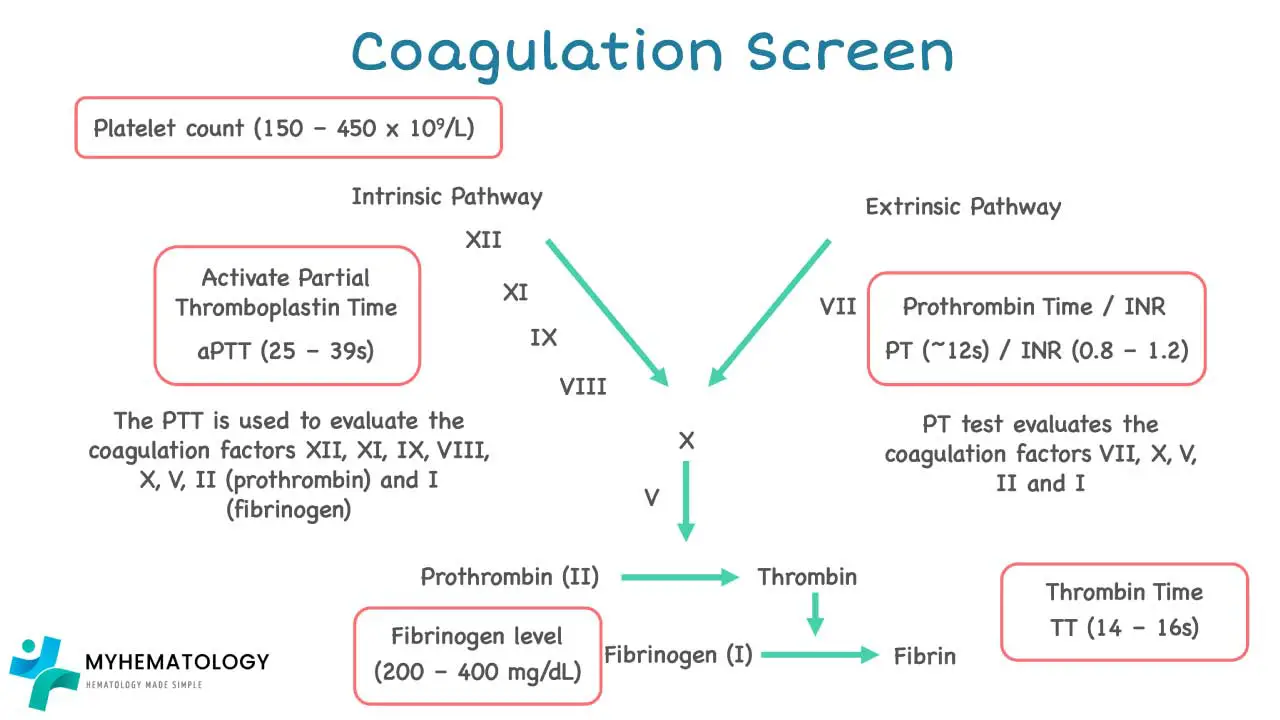
Fibrinogen Level
This test quantifies the amount of fibrinogen (Factor I) in the blood.
Reduced fibrinogen levels may be due to consumption (as seen in DIC), liver impairment, hemodilution, or congenital deficiency. Fibrinogen is also an acute phase reactant, meaning its levels can rise in response to inflammation, infection, malignancy, and pregnancy.
Activated Clotting Time (ACT)
The ACT is a rapid, point-of-care test primarily used to monitor high doses of unfractionated heparin therapy. It measures the inhibiting effect of heparin on the clotting system, rather than the actual heparin level.
- Normal Range: Typically 80-140 seconds.
It is commonly used during medical or surgical procedures that require significant anticoagulation, such as heart bypass surgery, coronary angioplasty, dialysis, and extracorporeal membrane oxygenation (ECMO). The ACT can be influenced by platelet count and function, blood temperature, and underlying coagulation factor deficiencies.
This leads to higher perceived value and authority, translating to improved rankings due to increased user satisfaction and relevance.
Coagulation Factors and Their Associated Bleeding or Thrombotic Disorders
| Factor Number | Common Name | Pathway(s) Involved | Primary Function | Associated Bleeding/Thrombotic Disorders |
| I | Fibrinogen | Common | Precursor to fibrin; forms clot mesh | Dysfibrinogenemia, Afibrinogenemia, Hypofibrinogenemia |
| II | Prothrombin | Common | Precursor to thrombin; activates fibrinogen | Prothrombin deficiency, Prothrombin gene mutation |
| V | Labile Factor | Common | Cofactor for Factor Xa; accelerates thrombin formation | Factor V deficiency, Factor V Leiden thrombophilia |
| VII | Stable Factor | Extrinsic | Initiates extrinsic pathway with Tissue Factor | Factor VII deficiency |
| VIII | Antihemophilic Factor | Intrinsic, Common (Cofactor) | Cofactor for Factor IXa; accelerates Factor X activation | Hemophilia A |
| IX | Christmas Factor | Intrinsic | Activates Factor X in intrinsic pathway | Hemophilia B |
| X | Stuart-Prower Factor | Extrinsic, Intrinsic, Common | Activates Factor II (prothrombin) | Factor X deficiency |
| XI | Plasma Thromboplastin Antecedent | Intrinsic | Activates Factor IX | Factor XI deficiency |
| XII | Hageman Factor | Intrinsic | Initiates intrinsic pathway (less critical in vivo) | Factor XII deficiency (usually asymptomatic) |
| XIII | Fibrin-stabilizing Factor | Common | Cross-links fibrin to stabilize clot | Factor XIII deficiency |
| – | von Willebrand factor (vWF) | Primary Hemostasis, Intrinsic (carrier for FVIII) | Platelet adhesion; carries Factor VIII | von Willebrand Disease |
Specialized Coagulation Tests
When screening tests yield abnormal results, or specific conditions are suspected, more specialized tests are performed.
Coagulation Factor Assays
These are specific blood tests that check individual clotting factors to determine if there is too much, too little, a missing, or a non-functional clotting factor.
They are used to pinpoint the exact cause of a bleeding or clotting disorder, especially when PT/INR or APTT results are abnormal, or if there’s a family history of clotting factor problems. For example, Factor V assay measures Factor V, and abnormal levels may indicate liver disease or DIC. These assays can identify specific inherited bleeding disorders like hemophilia.
Mixing Studies (Correction Studies)
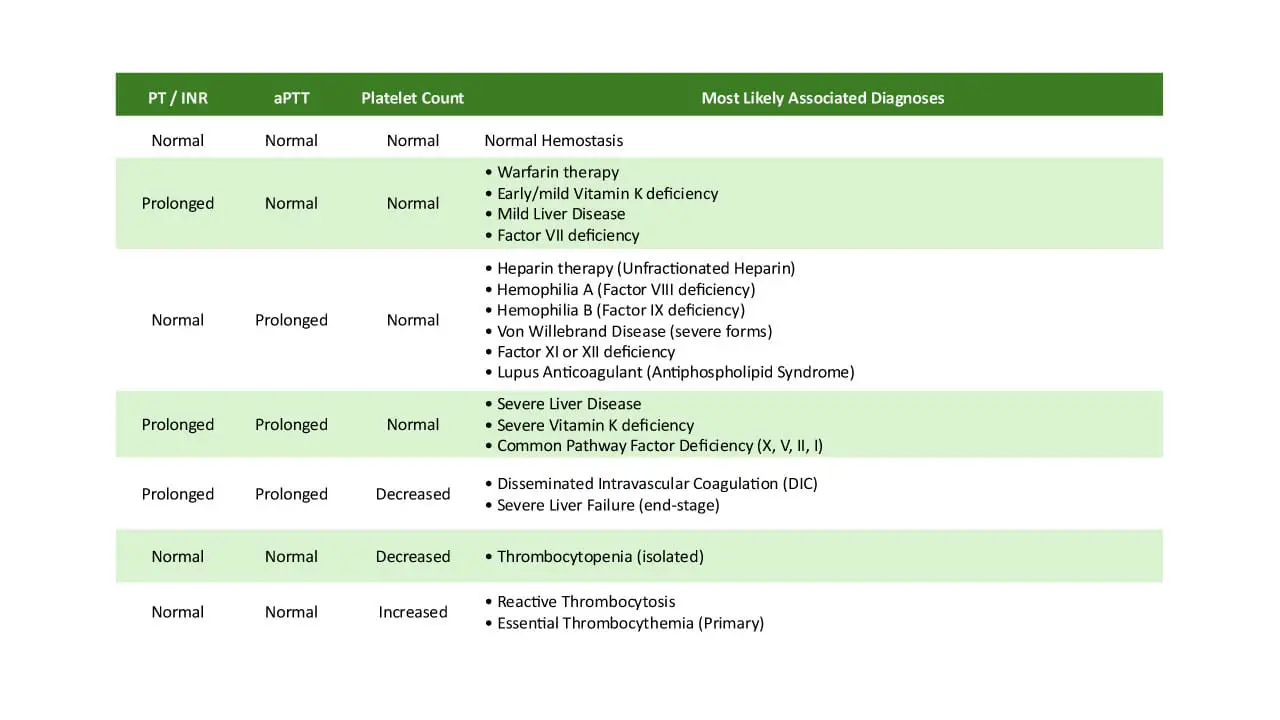
Mixing studies are performed when PT/INR and/or APTT results are prolonged. The patient’s plasma is mixed 50:50 with normal pooled plasma (NPP), and the PT/INR or APTT is re-measured.
Correction: If the prolonged PT/INR or APTT normalizes after mixing with NPP, it suggests a factor deficiency. This is because the NPP provides the missing or deficient clotting factors. Common causes include Vitamin K deficiency, warfarin therapy, congenital factor deficiencies, liver failure, or DIC.
Non-Correction: If the prolonged PT/INR or APTT remains elevated despite the addition of NPP, it indicates the presence of an inhibitor in the patient’s plasma. This inhibitor is neutralizing the clotting factors in both the patient’s and the normal plasma. Examples of inhibitors include heparin, direct thrombin or Factor Xa inhibitors, lupus anticoagulant, or specific factor antibodies.
Summary of Laboratory Investigations Related to the Coagulation Cascade
| Test Name | Pathways Evaluated | What it Measures | Clinical Utility/Significance | Common Conditions Causing Abnormal Results |
| Prothrombin Time (PT) & INR | Extrinsic & Common | Time for plasma to clot after adding thromboplastin | Monitors warfarin therapy; screens for liver disease, Vitamin K deficiency, Factor VII deficiency | Warfarin therapy, Vitamin K deficiency, Liver disease, Factor VII, X, V, II, I deficiencies |
| Activated Partial Thromboplastin Time (APTT) | Intrinsic & Common | Time for plasma to clot after adding activator & phospholipid | Monitors heparin therapy; screens for hemophilia, von Willebrand disease, Factor XII, XI, IX, VIII deficiencies | Heparin therapy, Hemophilia A/B, von Willebrand disease (severe), DIC, Factor XII, XI, IX, VIII, X, V, II, I deficiencies |
| Thrombin Time (TT) | Common | Time for fibrinogen to convert to fibrin | Assesses fibrinogen function/concentration; detects heparin contamination | Heparin contamination, Hypofibrinogenemia, Dysfibrinogenemia |
| Fibrinogen Level | Common | Concentration of fibrinogen in plasma | Assesses clotting ability; indicator in inflammatory states | DIC, Liver disease, Afibrinogenemia (low); Inflammation (high) |
| Activated Clotting Time (ACT) | Global | Overall clotting time at point-of-care | Monitors high-dose heparin during procedures (e.g., cardiac surgery) | High-dose heparin |
| Coagulation Factor Assays | Specific Factors | Activity level of individual clotting factors | Diagnoses specific factor deficiencies (e.g., Hemophilia) | Specific factor deficiencies |
| Mixing Studies (Correction Studies) | Intrinsic & Extrinsic | Differentiates factor deficiency from inhibitor | Helps determine if prolonged PT/APTT is due to factor deficiency or inhibitor | Factor deficiency (corrects), Inhibitor (does not correct) |
General Treatment and Management
Understanding the intricate steps of the coagulation cascade is crucial for pinpointing the specific factor responsible for a bleeding disorder. By analyzing the specific stage of the coagulation cascade where clotting fails, doctors can diagnose the underlying defect and recommend appropriate treatment. This might involve:
- Replacement therapy: Infusing the missing factor back into the patient’s blood, enabling proper clot formation.
- Medications: Drugs that stimulate the production of certain factors or bypass the defective step in the cascade.
- Blood products: Transfusions with platelets or plasma concentrates can provide temporary support for clotting.
While treatment remains important, understanding the coagulation cascade also plays a vital role in preventing unwanted clotting. In conditions like deep vein thrombosis or pulmonary embolism, where clots form within blood vessels, anticoagulant medications come into play. These drugs target specific factors in the coagulation cascade, like thrombin or factor Xa, to inhibit their activity and prevent clot formation.
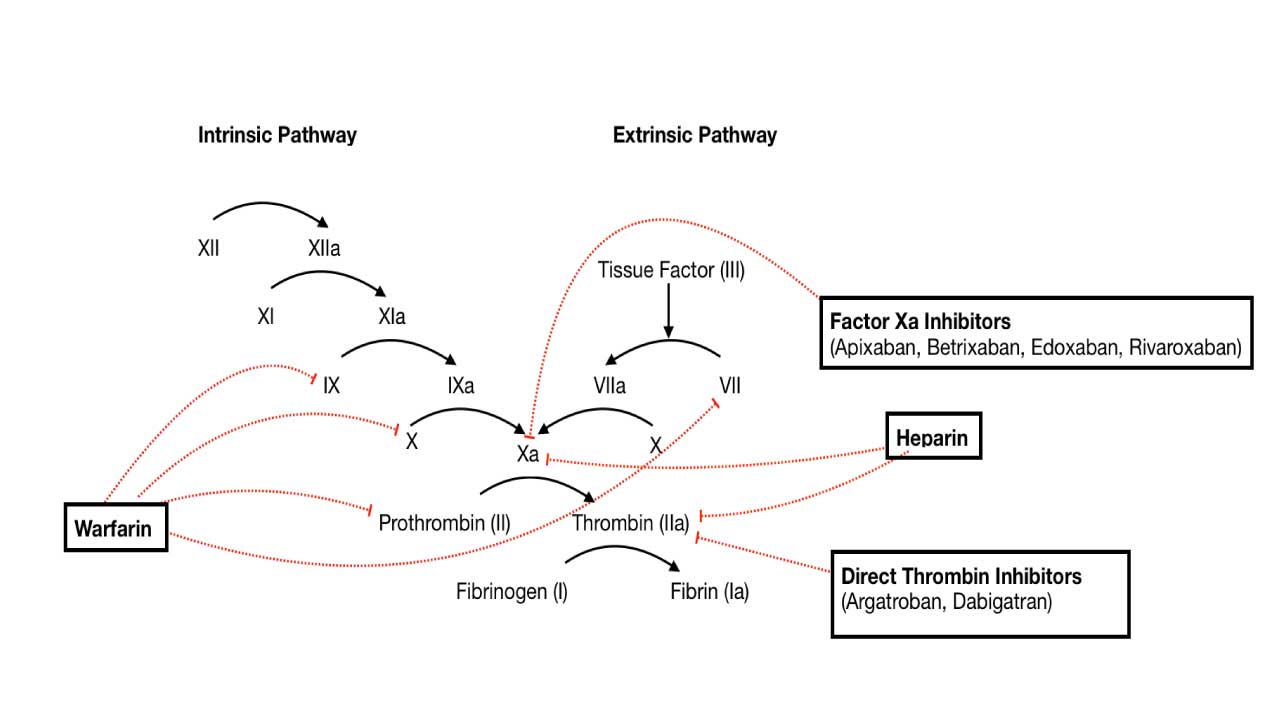
Pharmacological Modulation of Coagulation Cascade
Pharmacological modulation of the coagulation cascade is a critical aspect of modern medicine, allowing clinicians to either promote clotting (with procoagulants) or inhibit it (with anticoagulants) depending on the patient’s clinical needs. This intervention is essential for managing a wide range of conditions, from preventing life-threatening thrombosis to controlling severe bleeding.
Anticoagulants
Anticoagulants are medications designed to prevent or reduce blood clot formation by interfering with various steps of the coagulation cascade. They are widely used in conditions like deep vein thrombosis (DVT), pulmonary embolism (PE), atrial fibrillation, and after certain surgeries to prevent thrombotic events.
Heparin and Heparin-like Drugs (e.g., Fondaparinux)
These drugs enhance the activity of Antithrombin, a natural anticoagulant protein. Antithrombin primarily inactivates thrombin (Factor IIa) and Factor Xa, as well as Factors XIa and XIIa. By accelerating antithrombin’s action, heparin and fondaparinux effectively inhibit the propagation of the coagulation cascade.
Unfractionated heparin (UFH) is used for rapid anticoagulation in acute thrombotic events, during surgical procedures requiring significant anticoagulation (like cardiopulmonary bypass), and to monitor high doses of heparin therapy using the Activated Clotting Time (ACT) test.
The activated Partial Thromboplastin Time (APTT) is used to monitor regular-dose unfractionated heparin. Fondaparinux is a synthetic pentasaccharide that selectively inhibits Factor Xa via antithrombin.
Vitamin K Antagonists (e.g., Warfarin)
Warfarin interferes with the liver’s ability to synthesize functional Vitamin K-dependent coagulation factors: Factor II (prothrombin), Factor VII, Factor IX, and Factor X, as well as the natural anticoagulant proteins Protein C and Protein S. These factors require Vitamin K for a post-translational modification (gamma-carboxylation) that enables them to bind calcium and participate in the coagulation cascade. By inhibiting this process, warfarin leads to the production of inactive clotting factors.
Warfarin is a long-term oral anticoagulant used to prevent blood clots in conditions such as atrial fibrillation, prosthetic heart valves, and recurrent DVT/PE. Its effect is monitored using the Prothrombin Time (PT) and its standardized derivative, the International Normalized Ratio (INR).
Direct Oral Anticoagulants (DOACs)
- Direct Thrombin Inhibitors (DTIs) / Factor IIa Inhibitors (e.g., Dabigatran, Argatroban, Bivalirudin): These drugs directly bind to and inhibit thrombin (Factor IIa), preventing its ability to convert fibrinogen to fibrin and activate other factors in the coagulation cascade. Dabigatran is an oral DTI used for stroke prevention in atrial fibrillation and DVT/PE treatment. Argatroban and bivalirudin are parenteral DTIs used in specific clinical settings, such as heparin-induced thrombocytopenia (HIT) or during percutaneous coronary intervention (PCI). The Thrombin Time (TT) can be prolonged by these drugs.
- Direct Factor Xa Inhibitors (e.g., Rivaroxaban, Apixaban, Edoxaban): These drugs directly inhibit Factor Xa, thereby preventing the formation of thrombin from prothrombin. Widely used for stroke prevention in atrial fibrillation, and for the treatment and prevention of DVT and PE. They generally do not require routine coagulation monitoring.
Procoagulants
Procoagulants are agents used to promote blood clotting and stop bleeding, particularly in patients with bleeding disorders or those experiencing excessive blood loss.
Coagulation Factor Concentrates
These products directly replace deficient or consumed coagulation factors, thereby restoring the body’s ability to form a clot.
- Clinical Use:
- Factor VIII Concentrates: Used to treat Hemophilia A.
- Factor IX Concentrates: Used to treat Hemophilia B.
- Prothrombin Complex Concentrates (PCCs): Contain varying amounts of Vitamin K-dependent factors (II, VII, IX, X) and sometimes Protein C and Protein S. They are used to rapidly reverse the effects of Vitamin K antagonists (like warfarin) in cases of major bleeding or urgent surgery, or to treat deficiencies of the factors they contain.
- Cryoprecipitate: Rich in Factor VIII, von Willebrand factor, Factor XIII, and fibrinogen. Used to replace fibrinogen in cases of severe hypofibrinogenemia (low fibrinogen levels) or to treat von Willebrand disease.
- Fresh Frozen Plasma (FFP): Contains all coagulation factors, inhibitors, and albumin. Used to replace multiple clotting factors in patients with liver disease, DIC, or massive transfusion, when specific factor concentrates are not available or appropriate.
- Recombinant Activated Human Factor VII (rFVIIa): A potent procoagulant that directly activates Factor X on the surface of activated platelets, bypassing the intrinsic and extrinsic pathways of the coagulation cascade. Used in specific situations for severe bleeding in patients with inhibitors to Factor VIII or IX, or in certain types of trauma.
Desmopressin (DDAVP)
Desmopressin is a synthetic analog of vasopressin that primarily acts by increasing the release of von Willebrand factor (vWF) and Factor VIII from endothelial cells. vWF is crucial for platelet adhesion and also stabilizes Factor VIII.
It is used to improve platelet function and promote clotting in patients with mild Hemophilia A or certain types of von Willebrand disease, and sometimes for uremic bleeding.
Topical Hemostatic Agents
These agents are applied directly to the site of bleeding to promote localized clot formation.
- Clinical Use:
- Thrombin and Fibrin Glue: Used surgically to treat localized bleeding and to thrombose aneurysms. Fibrin glue contains fibrinogen and thrombin, which rapidly form a fibrin clot upon mixing.
- Adsorbent Chemicals (e.g., Zeolites) and Hemostatic Powder Sprays (e.g., TC-325): Used for sealing severe injuries quickly, such as in traumatic bleeding or to treat gastrointestinal bleeding. They work by concentrating clotting factors or providing a matrix for clot formation.
Vitamin K
As mentioned, Vitamin K is essential for the synthesis of functional Factors II, VII, IX, X, Protein C, and Protein S.
It is administered to reverse the effects of warfarin (Vitamin K antagonist) or to treat Vitamin K deficiency, which can cause bleeding due to impaired clotting factor synthesis.
The careful selection and monitoring of these pharmacological agents are vital to maintain hemostatic balance, preventing both excessive bleeding and dangerous thrombosis in patients.
Frequently Asked Questions (FAQs)
How does warfarin affect the coagulation cascade?
Warfarin is an anticoagulant medication that works by interfering with vitamin K, a crucial cofactor for several proteins involved in the coagulation cascade.
Mechanism of Action
Warfarin inhibits the enzyme vitamin K epoxide reductase, which is responsible for recycling vitamin K after its use in the synthesis of specific clotting factors. By blocking this enzyme, warfarin effectively depletes the body’s supply of active vitamin K.
Impact on Coagulation Factors
Vitamin K is essential for the synthesis of several key clotting factors, including:
- Factors II, VII, IX, and X: These factors are directly involved in the coagulation cascade, leading to thrombin formation and fibrin clot formation.
- Proteins C and S: These are natural anticoagulants that help regulate the clotting process.
By inhibiting vitamin K, warfarin indirectly reduces the levels of these clotting factors, leading to a prolonged prothrombin time (PT) and an increased risk of bleeding.
How come the extrinsic and intrinsic pathways are named that way?
The names “extrinsic” and “intrinsic” for the two arms of the coagulation cascade are derived from the way blood clotting was historically measured in laboratory tests, rather than how the process works within the body.
- Extrinsic Pathway: This pathway is so named in the coagulation cascade because to initiate clotting in a test tube, a substance “extrinsic” (meaning “from outside”) to the blood itself must be added. This substance is tissue factor, which is not normally found circulating freely in the blood. The prothrombin time (PT) and International Normalized Ratio (INR) tests specifically measure this pathway by adding a tissue factor-like substance to the blood sample.
- Intrinsic Pathway: This pathway is called “intrinsic” in the coagulation cascade because all the components needed for its activation are already “intrinsic” (meaning “from within”) to the blood itself. No external substance needs to be added to the test tube; simply exposing the blood to a negatively charged surface, such as glass, is enough to start the process. The activated partial thromboplastin time (APTT) test measures this pathway.
Which factor initiates the coagulation cascade in vivo?
The coagulation cascade in the human body (in vivo) is primarily initiated by tissue factor (TF).
When a blood vessel is damaged, tissue factor, which is normally located in the subendothelial layer of the vessel wall, is exposed to the bloodstream. This exposure activates factor VII (FVII) into its active form, factor VIIa, forming a complex that is the primary initiator of coagulation.
Why do the PT/INR and PTT measure different pathways?
The Prothrombin Time (PT) and Activated Partial Thromboplastin Time (APTT or PTT) measure different coagulation cascade pathways because they were specifically designed in a laboratory setting to assess different parts of the clotting process. The naming of the pathways, extrinsic and intrinsic, in the coagulation cascade is a direct result of the specific reagents added to a blood sample in a test tube.
- Prothrombin Time (PT) and INR: The PT and its standardized form, the International Normalized Ratio (INR), are designed to measure the extrinsic pathway and the final common pathway of the coagulation cascade. To perform this test, a substance called thromboplastin, which acts like tissue factor, is added to the blood sample. Since tissue factor is a component “extrinsic” to the normal circulation, it activates the extrinsic pathway, and the time it takes for a clot to form is measured.
- Activated Partial Thromboplastin Time (APTT or PTT): The PTT measures the intrinsic pathway of the coagulation cascade. This test is initiated by adding a reagent that mimics a negatively charged surface, such as glass, to the blood sample. All the factors needed to start this pathway are already “intrinsic” to the blood itself, so nothing “extrinsic” to the blood is needed to begin the clotting process.
Disclaimer: This article is intended for informational purposes only and is specifically targeted towards medical students. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Saba HI, Roberts HR. Hemostasis and Thrombosis: Practical Guidelines in Clinical Management (Wiley Blackwell). 2014.
- DeLoughery TG. Hemostasis and Thrombosis 4th Edition (Springer). 2019.
- Keohane EM, Otto CN, Walenga JM. Rodak’s Hematology 6th Edition (Saunders). 2019.
- Kaushansky K, Levi M. Williams Hematology Hemostasis and Thrombosis (McGraw-Hill). 2017.
- Starikova, E. A., Mammedova, J. T., Rubinstein, A. A., Sokolov, A. V., & Kudryavtsev, I. V. (2025). Activation of the Coagulation Cascade as a Universal Danger Sign. Current Issues in Molecular Biology, 47(2), 108. https://doi.org/10.3390/cimb47020108
- Park S, Park JK. Back to basics: the coagulation pathway. BR 2024;59:-. https://doi.org/10.1007/s44313-024-00040-8
- Chaudhry R, Killeen RB, Babiker HM. Physiology, Coagulation Pathways. [Updated 2025 Jun 2]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482253/
- New insight into the traditional model of the coagulation cascade and its regulation: illustrated review of a three-dimensional viewTroisi, Romualdo et al.Research and Practice in Thrombosis and Haemostasis, Volume 7, Issue 6, 102160
- Al-Koussa, H., AlZaim, I., & El-Sabban, M. E. (2022). Pathophysiology of Coagulation and Emerging Roles for Extracellular Vesicles in Coagulation Cascades and Disorders. Journal of Clinical Medicine, 11(16), 4932. https://doi.org/10.3390/jcm11164932
- Toh CH, Yong J. Rethinking coagulation: from enzymatic cascade and cell-based reactions to a convergent model involving innate immune activation. Blood (2023) 142 (25): 2133–2145. https://doi.org/10.1182/blood.2023021166
- Adams, R. L., & Bird, R. J. (2009). Review article: Coagulation cascade and therapeutics update: relevance to nephrology. Part 1: Overview of coagulation, thrombophilias and history of anticoagulants. Nephrology (Carlton, Vic.), 14(5), 462–470. https://doi.org/10.1111/j.1440-1797.2009.01128.x
- Raber MN. Coagulation Tests. In: Walker HK, Hall WD, Hurst JW, editors. Clinical Methods: The History, Physical, and Laboratory Examinations. 3rd edition. Boston: Butterworths; 1990. Chapter 157. Available from: https://www.ncbi.nlm.nih.gov/books/NBK265/