Key Takeaways
Evans syndrome is. a rare autoimmune disorder with sequential or simultaneous autoimmune hemolytic anemia (AIHA) and immune thrombocytopenia (ITP), sometimes with autoimmune neutropenia (AIN).
- Pathophysiology ▾: Immune dysregulation leads to autoantibodies targeting red blood cells, platelets, and/or neutrophils, causing their destruction. Genetic predispositions are increasingly recognized.
- Symptoms ▾: Variable, including fatigue, pallor, bruising, bleeding, jaundice, and potential for infections if neutropenia is present.
- Investigations ▾: Diagnosis relies on demonstrating AIHA (positive DAT, hemolysis), ITP (low platelets, exclusion), and/or AIN (low neutrophils, exclusion), alongside ruling out secondary causes.
- Treatment ▾: Aims to control autoimmune destruction with corticosteroids, IVIG, rituximab, splenectomy, immunosuppressants, and supportive care (transfusions).
- Potential Complications ▾: Severe anemia, bleeding, infections, treatment side effects, and potential for other autoimmune disorders or, rarely, thromboembolic events.
*Click ▾ for more information
Introduction
Evans syndrome is a rare autoimmune disorder characterized by the sequential or simultaneous occurrence of at least two of the following immune-mediated cytopenias (blood cell deficiencies):
- Autoimmune Hemolytic Anemia (AIHA)
- Immune Thrombocytopenia (ITP)
- Autoimmune Neutropenia (AIN)
Evans syndrome can be challenging to diagnose due to its rarity and the variability in its presentation. The sequential or overlapping nature of the cytopenias can make it difficult to distinguish from other autoimmune disorders or hematologic conditions. Accurate diagnosis is crucial for appropriate management.
Causes and Pathophysiology of Evans Syndrome
Evans syndrome is a complex autoimmune disorder, and while the exact causes are not fully understood in many cases (idiopathic Evans syndrome), current research points to a combination of immune dysregulation and, in some instances, identifiable underlying factors.
Causes
- Idiopathic (Primary) Evans Syndrome: In the majority of cases, the trigger for the autoimmune attack remains unknown. It's believed to arise from a fundamental problem in immune regulation, where the body's tolerance to its own blood cells is lost.
- Secondary Evans Syndrome: In a significant subset of patients, Evans syndrome can be associated with or secondary to other underlying conditions.
- Other Autoimmune Diseases: Systemic Lupus Erythematosus (SLE), Antiphospholipid Syndrome, Sjögren's syndrome, and Autoimmune Lymphoproliferative Syndrome (ALPS) are known associations.
- Lymphoproliferative Disorders: Conditions like Chronic Lymphocytic Leukemia (CLL) and Non-Hodgkin Lymphoma can sometimes precede or occur concurrently with Evans syndrome.
- Primary Immunodeficiencies: Common Variable Immunodeficiency (CVID) and Selective IgA Deficiency have been linked to an increased risk of developing Evans syndrome.
- Infections: Viral infections (e.g., HIV, Hepatitis C) have been implicated as potential triggers in some cases.
- Genetic Predisposition: While generally considered sporadic, familial occurrences are rare. Emerging research highlights that a significant proportion of children with Evans syndrome (up to 65% in some studies) may have underlying genetic variants affecting immune regulation. These variants can involve genes crucial for lymphocyte apoptosis (like FAS in ALPS), immune signaling pathways (like CTLA4, LRBA, STAT3, PIK3CD), and other aspects of immune homeostasis. Identifying these genetic causes can have prognostic significance and potentially guide targeted treatment.
Pathophysiology
The central mechanism in Evans syndrome involves a breakdown of immune tolerance, leading to the production of autoantibodies that target the body's own blood cells.
- Autoantibody Production: B lymphocytes mistakenly produce antibodies against antigens on the surface of red blood cells, platelets, and sometimes neutrophils.
- In Autoimmune Hemolytic Anemia (AIHA), these autoantibodies, predominantly of the IgG "warm" type, bind to red blood cells. These antibody-coated red blood cells are then prematurely destroyed, primarily by phagocytic cells in the spleen (extravascular hemolysis). Complement activation (intravascular hemolysis) can also occur but is less common in warm AIHA associated with Evans syndrome. The Direct Antiglobulin Test (DAT or Coombs test) is positive, detecting these antibodies or complement components on the red blood cell surface.
- In Immune Thrombocytopenia (ITP), autoantibodies (often targeting glycoproteins like GPIIb/IIIa on the platelet surface) lead to increased platelet destruction, mainly in the spleen. Additionally, autoantibodies can impair megakaryocyte function, reducing platelet production.
- In Autoimmune Neutropenia (AIN), autoantibodies target neutrophil-specific antigens, leading to their premature destruction.
- T-Cell Dysregulation: While autoantibodies play a direct role in cell destruction, dysregulation of T cells (another type of immune cell) is also thought to be crucial in the pathogenesis of Evans syndrome. Imbalances between helper T cells and suppressor T cells, as well as defects in T-cell mediated suppression of autoreactive B cells, likely contribute to the development and persistence of the autoimmune response.
- Impaired Apoptosis: In some cases, particularly those associated with ALPS, defects in lymphocyte apoptosis (programmed cell death) can lead to an accumulation of autoreactive lymphocytes that would normally be eliminated. This failure to remove self-reactive immune cells contributes to the development of autoimmunity.
- Molecular Mimicry and Bystander Activation: While less clearly defined in Evans syndrome, it's hypothesized that infections might trigger the production of autoantibodies through molecular mimicry (where microbial antigens resemble self-antigens) or bystander activation (where inflammation from an infection nonspecifically activates autoreactive immune cells).
Signs and Symptoms of Evans Syndrome
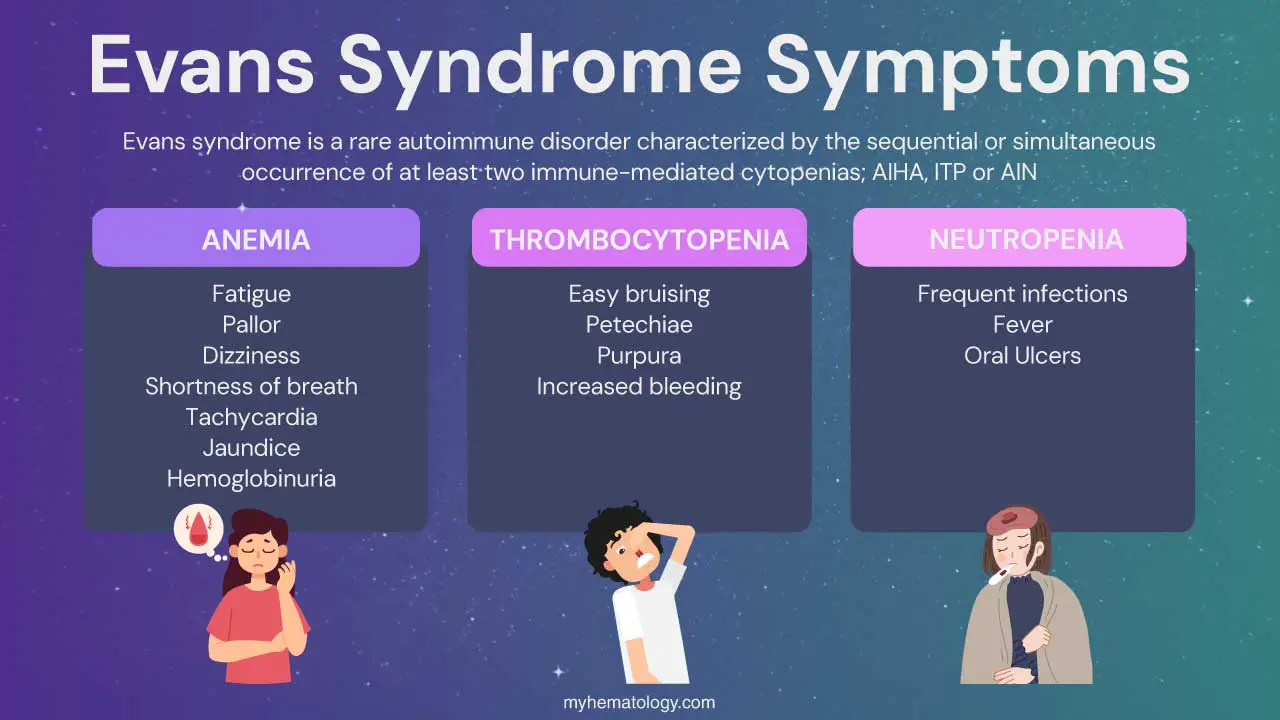
The signs and symptoms of Evans syndrome are variable and depend on which blood cell lines are affected. Patients may experience symptoms related to low red blood cells (anemia), low platelets (thrombocytopenia), and less commonly, low neutrophils (neutropenia). These symptoms can occur simultaneously or sequentially.
Symptoms related to low red blood cells (anemia)
- Fatigue and weakness
- Pallor
- Dizziness and lightheadedness
- Shortness of breath (dyspnea)
- Rapid heartbeat (tachycardia) and palpitations
- Jaundice
- Dark urine (hemoglobinuria)
Symptoms related to low platelets (thrombocytopenia)
- Easy bruising (ecchymoses)
- Petechiae: Tiny red or purple spots on the skin, often resembling a rash, due to small bleeds under the skin.
- Purpura: Larger purple patches on the skin caused by bleeding under the surface.
- Increased bleeding: Prolonged bleeding from minor cuts, nosebleeds (epistaxis), bleeding gums, and heavy menstrual periods (menorrhagia).
- Rarely, severe bleeding: Including gastrointestinal bleeding or intracranial hemorrhage.
Symptoms related to low neutrophils (neutropenia)
- Frequent infections
- Fever
- Mouth sores (oral ulcers)
Other general signs and symptoms
- Splenomegaly
- Lymphadenopathy
- Hepatomegaly
The course of Evans syndrome is often characterized by periods of exacerbation (when symptoms worsen) and remission (when symptoms improve, often temporarily with treatment). The severity and combination of these signs and symptoms can vary greatly among individuals with Evans syndrome.
Laboratory Investigations of Evans Syndrome
The diagnosis of Evans syndrome is primarily based on clinical findings and the exclusion of other causes of the combined cytopenias. Laboratory investigations play a crucial role in confirming the presence of autoimmune hemolytic anemia (AIHA), immune thrombocytopenia (ITP), and/or autoimmune neutropenia (AIN), as well as in ruling out other conditions. There is no single definitive test for Evans syndrome itself.
Initial Hematological Assessment
- Complete Blood Count (CBC) with Differential: This is the cornerstone of the initial evaluation. It will reveal:
- Anemia
- Thrombocytopenia
- Neutropenia
- Peripheral Blood Smear: Examination of the blood smear can provide additional information:
- AIHA: May show abnormal red cell morphology like spherocytes (small, round red blood cells lacking central pallor), polychromasia (indicating increased young red blood cells/reticulocytes), and nucleated red blood cells (in severe hemolysis).
- ITP: Platelet morphology is usually normal, but large platelets (megathrombocytes or stress platelets) may be seen, indicating increased platelet production. The platelet count will be low. The smear helps rule out other causes of thrombocytopenia like thrombotic thrombocytopenic purpura (TTP) or disseminated intravascular coagulation (DIC).
- AIN: May show a reduced number of neutrophils with otherwise normal morphology.
- Reticulocyte Count: In AIHA, a high reticulocyte count indicates that the bone marrow is trying to compensate for the red blood cell destruction. A low reticulocyte count in the presence of anemia might suggest bone marrow failure or other underlying issues.
Confirmation of Autoimmune Hemolytic Anemia (AIHA)
- Direct Antiglobulin Test (DAT) or Coombs Test: This is crucial for diagnosing AIHA. It detects the presence of antibodies (IgG or IgM) and/or complement components (C3) on the surface of red blood cells. A positive DAT strongly supports the diagnosis of AIHA. The pattern of reactivity (e.g., IgG only, C3 only, or both) can sometimes provide clues about the type of AIHA (warm AIHA vs. cold agglutinin disease).
- Indirect Antiglobulin Test (IAT) or Indirect Coombs Test: This test detects the presence of free antibodies in the patient's plasma that can react with normal red blood cells. It may be positive in Evans syndrome, reflecting the presence of autoantibodies.
- Markers of Hemolysis
- Lactate Dehydrogenase (LDH): Elevated levels indicate cell damage, including red blood cell lysis.
- Bilirubin (Indirect/Unconjugated): Increased levels due to the breakdown of hemoglobin.
- Haptoglobin: Decreased levels as haptoglobin binds free hemoglobin released during hemolysis.
Assessment for Immune Thrombocytopenia (ITP)
Diagnosis of ITP in the context of Evans syndrome is largely one of exclusion after confirming thrombocytopenia and ruling out other causes of low platelets (e.g., drug-induced, infections, other systemic diseases).
- Anti-platelet Antibody Testing: While available, these tests are not highly sensitive or specific and are not routinely recommended for the diagnosis of ITP or Evans syndrome. A positive result can support the diagnosis, but a negative result does not rule it out.
Evaluation for Autoimmune Neutropenia (AIN)
Diagnosis of AIN is also often one of exclusion.
- Neutrophil Antibody Testing (e.g., Granulocyte Agglutination Test (GAT), Indirect Granulocyte Immunofluorescence Test (I-GIFT)): These tests detect antibodies against neutrophils. However, they have limitations in sensitivity and specificity and are not always readily available. Positive results can support the diagnosis, but negative results do not exclude AIN.
Investigations to Identify Underlying Causes (Secondary Evans Syndrome)
It's crucial to investigate for underlying conditions that may be associated with secondary Evans syndrome.
- Autoimmune Workup
- Antinuclear Antibody (ANA)
- Anti-dsDNA antibodies (for SLE)
- Antiphospholipid antibodies (lupus anticoagulant, anticardiolipin antibodies, anti-beta2-glycoprotein I antibodies)
- Rheumatoid Factor (RF)
- Anti-SSA/Ro and Anti-SSB/La antibodies (for Sjögren's syndrome)
- Serological Tests for Infections: HIV, Hepatitis C, EBV, cytomegalovirus (CMV), Parvovirus B19 (if clinically indicated).
- Immunoglobulin Levels (IgG, IgA, IgM): To screen for immunodeficiencies like common variable immunodeficiency (CVID).
- Lymphocyte Subset Analysis (Flow Cytometry): To evaluate T-cell and B-cell populations, which may be abnormal in certain associated conditions like ALPS or CVID.
- Genetic Testing: May be considered, especially in children with Evans syndrome, to identify underlying genetic variants associated with immune dysregulation (e.g., FAS gene in ALPS).
- Serum Protein Electrophoresis and Immunofixation: To look for monoclonal gammopathies associated with lymphoproliferative disorders.
Bone Marrow Examination
A bone marrow aspirate and biopsy are not always necessary for the diagnosis of classic Evans syndrome, especially when AIHA and ITP are clearly present with supportive laboratory findings. However, bone marrow examination may be indicated in certain situations.
In Evans syndrome, the bone marrow typically shows erythroid hyperplasia (increased red blood cell precursors) in AIHA, increased megakaryocytes (platelet precursors) in ITP, and normal or increased myeloid precursors.
Treatment and Management of Evans Syndrome
The treatment and management of Evans syndrome are challenging and often involve a multidisciplinary approach, primarily focusing on controlling acute episodes, preventing severe complications, and achieving sustained remission with minimal side effects. The approach is often tailored to the individual patient's specific symptoms, the severity of their condition, and the specific blood cell lines affected.
First-Line Treatment
- Corticosteroids (e.g., Prednisone): These are often the initial treatment of choice for both AIHA and ITP components of Evans syndrome. They suppress the immune system by reducing antibody production and decreasing the phagocytic activity of macrophages in the spleen. Long-term use can lead to significant side effects (weight gain, mood changes, hyperglycemia, osteoporosis, increased risk of infection), so the goal is to achieve remission with the lowest effective dose or transition to other agents.
- Intravenous Immunoglobulin (IVIG): This concentrated solution of pooled antibodies from healthy donors can be effective, particularly for acute ITP and sometimes AIHA. The response is often transient, and it's primarily used for rapid platelet or red blood cell count improvement in critical situations. Side effects are generally mild but can include headache, fever, and chills.
Second-Line Treatment (for Refractory or Relapsing Disease)
These options are considered when first-line therapies fail to achieve a sustained response or when patients experience frequent relapses or significant steroid-related side effects.
- Rituximab: A monoclonal antibody that targets the CD20 protein on B cells, leading to their depletion. It can be effective in a significant proportion of patients, but responses may not be durable. Potential side effects include infusion reactions and an increased risk of infection.
- Splenectomy: Surgical removal of the spleen, the primary site of red blood cell and platelet destruction. It can be effective in achieving remission in some patients, but it's an irreversible procedure with potential long-term risks, including an increased risk of overwhelming post-splenectomy infection (OPSI) by encapsulated bacteria. Patients should be vaccinated against Streptococcus pneumoniae, Haemophilus influenzae type b (Hib), and Neisseria meningitidis before or after splenectomy. Prophylactic antibiotics may be considered.
- Thrombopoietin Receptor Agonists (TPO-RAs): Eltrombopag, romiplostim, and avatrombopag primarily stimulate platelet production and are mainly used for the ITP component of Evans syndrome.
- Other Immunosuppressants
- Azathioprine: An antimetabolite that suppresses immune cell proliferation.
- Mycophenolate Mofetil (MMF): Inhibits purine synthesis, affecting lymphocyte proliferation.
- Cyclosporine A: Inhibits T-cell activation.
- Sirolimus (Rapamycin): Inhibits the mTOR pathway, affecting T-cell proliferation and function.
- Considerations: These agents have varying degrees of efficacy in Evans syndrome and are often used as steroid-sparing agents or for patients refractory to other treatments. They have their own potential side effects that require careful monitoring.
- Alemtuzumab: A monoclonal antibody targeting CD52, leading to the depletion of both B and T lymphocytes. Reserved for very refractory cases due to significant immunosuppression and potential for severe infections.
Supportive Care
- Blood Transfusions (Packed Red Blood Cells): Used to treat severe symptomatic anemia. Transfusions are generally reserved for situations where the patient is significantly symptomatic due to the risk of alloimmunization (development of antibodies against donor red blood cells).
- Platelet Transfusions: Used for severe bleeding episodes or before invasive procedures in patients with very low platelet counts. Platelet transfusions may have limited efficacy due to ongoing immune destruction.
- Management of Infections: Prompt diagnosis and treatment of infections are crucial, especially in patients with neutropenia or those on immunosuppressive medications. Prophylactic antibiotics or antifungals may be considered in some cases.
- Psychological Support: Living with a chronic and unpredictable autoimmune disorder can be emotionally challenging. Providing psychological support, counseling, and connecting patients with support groups can be beneficial.
Prognosis and Potential Complications
The prognosis of Evans syndrome is variable and often unpredictable. While some individuals may achieve periods of remission, the disease is frequently chronic with relapsing and remitting episodes. The potential for complications depends on the severity and frequency of these episodes, the specific blood cell lines affected, and the intensity and duration of treatment.
Prognosis
For many patients, Evans syndrome follows a chronic course characterized by recurrent episodes of anemia, thrombocytopenia, or neutropenia. These relapses can occur spontaneously or be triggered by infections or other factors.
The response to various treatments can differ significantly among individuals. Some patients may achieve long-lasting remissions with first- or second-line therapies, while others may have more refractory disease requiring more aggressive or experimental approaches.
In cases of secondary Evans syndrome, the prognosis may be influenced by the underlying autoimmune disorder, lymphoproliferative disease, or immunodeficiency. Effective management of the primary condition can sometimes improve the course of Evans syndrome.
While advancements in diagnosis and treatment have improved outcomes, Evans syndrome can still be associated with increased morbidity and, in some cases, mortality. Causes of death can include severe bleeding, overwhelming infections, and complications from aggressive immunosuppressive therapies.
Potential Complications
The complications of Evans syndrome arise primarily from the severe cytopenias and the treatments used to manage the condition.
Complications Related to Severe Anemia (AIHA)
- Cardiac Complications: Chronic or severe anemia can lead to heart failure, arrhythmias, and angina due to the increased workload on the heart to compensate for reduced oxygen-carrying capacity.
- Severe Fatigue and Debilitation: Profound anemia can significantly impair daily activities and overall quality of life.
- Growth Retardation and Developmental Delays (in children): Chronic anemia can affect growth and development in pediatric patients.
Complications Related to Severe Thrombocytopenia (ITP)
- Severe Bleeding: The most serious risk of low platelet counts is significant bleeding, which can occur internally (e.g., gastrointestinal hemorrhage, intracranial hemorrhage) or externally. Intracranial hemorrhage, although rare, is a life-threatening complication.
- Increased Risk of Bleeding During Procedures: Even minor surgical or dental procedures can carry a higher risk of bleeding.
Complications Related to Severe Neutropenia (AIN)
- Overwhelming Infections: A low neutrophil count impairs the body's ability to fight off bacterial and fungal infections, which can be severe and life-threatening.
Complications Related to Treatment
- Corticosteroid Side Effects: Long-term use of corticosteroids can lead to a wide range of adverse effects, including weight gain, mood changes, osteoporosis, hypertension, diabetes, increased risk of infections, cataracts, and glaucoma.
- Immunosuppressant Side Effects: Other immunosuppressive medications can also have significant side effects, such as increased risk of infections, liver or kidney toxicity, gastrointestinal disturbances, and potential for malignancy with long-term use.
- Post-Splenectomy Complications: Splenectomy increases the risk of overwhelming post-splenectomy infection (OPSI) with encapsulated bacteria. There is also a potential for increased risk of thromboembolic events.
- Complications of Blood and Platelet Transfusions: These can include allergic reactions, transfusion-related acute lung injury (TRALI), transfusion-associated circulatory overload (TACO), and alloimmunization (development of antibodies against donor blood products), which can make future transfusions less effective.
- Increased Risk of Secondary Malignancies: Some immunosuppressive therapies have been associated with a slightly increased risk of certain cancers over the long term.
Other Potential Complications
- Development of Other Autoimmune Disorders: Individuals with Evans syndrome may have an increased risk of developing other autoimmune diseases over time.
- Thromboembolic Events: Although less common, some patients with Evans syndrome, particularly those with associated autoimmune conditions like antiphospholipid syndrome or after splenectomy, may have an increased risk of blood clots.
Frequently Asked Questions (FAQs)
What triggers Evans syndrome?
The precise triggers of Evans syndrome often remain elusive, particularly in cases of idiopathic (primary) Evans syndrome. However, current understanding suggests a complex interplay of factors leading to a fundamental breakdown in immune tolerance. In many instances, no single, identifiable trigger is apparent.
However, in a subset of patients with secondary Evans syndrome, certain underlying conditions are strongly associated and may act as precipitating or contributing factors. These include other autoimmune diseases like systemic lupus erythematosus (SLE) or antiphospholipid syndrome, lymphoproliferative disorders such as chronic lymphocytic leukemia (CLL), and primary immunodeficiencies like common variable immunodeficiency (CVID). Viral infections have also been implicated in some cases, potentially through mechanisms like molecular mimicry or bystander activation of the immune system. Furthermore, emerging research highlights a potential role for underlying genetic predispositions, with an increasing number of genetic variants affecting immune regulation being identified in individuals with Evans syndrome, suggesting that a genetic susceptibility might make certain individuals more prone to developing the condition when exposed to other environmental or internal triggers.
What is Evans syndrome's life expectancy?
The life expectancy for individuals with Evans syndrome is variable and influenced by several factors, including whether the condition is primary (idiopathic) or secondary to another underlying disease.
A nationwide study on Evans syndrome indicated a median survival of 7.2 years. However, this figure varies significantly between primary and secondary Evans syndrome. The median survival for primary Evans syndrome was notably longer at 10.9 years, while those with secondary Evans syndrome had a considerably shorter median survival of only 1.7 years. The five-year survival rate for secondary Evans syndrome in this study was reported to be around 38%.
The leading causes of death in individuals with Evans syndrome are reported to be bleeding, infections, and hematological cancer. Therefore, careful management focusing on preventing and treating these complications is crucial in improving the life expectancy of patients with Evans syndrome.
Is Evans syndrome congenital?
While Evans syndrome is generally considered an acquired autoimmune disorder and not typically congenital (present at birth), emerging research has identified underlying genetic variants affecting immune regulation in a significant proportion of patients, particularly children. These genetic factors may predispose individuals to develop Evans syndrome, suggesting a potential for a genetic susceptibility rather than the condition being directly inherited or present from birth in all cases. Familial occurrences of Evans syndrome are rare, but the increasing identification of these genetic underpinnings indicates that in some instances, a congenital predisposition to immune dysregulation plays a role in the development of the syndrome later in life.
Disclaimer: This article is intended for informational purposes only and is specifically targeted towards medical students. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Fattizzo B, Michel M, Giannotta JA, Hansen DL, Arguello M, Sutto E, Bianchetti N, Patriarca A, Cantoni S, Mingot-Castellano ME, McDonald V, Capecchi M, Zaninoni A, Consonni D, Vos JM, Vianelli N, Chen F, Glenthøj A, Frederiksen H, González-López TJ, Barcellini W. Evans syndrome in adults: an observational multicenter study. Blood Adv. 2021 Dec 28;5(24):5468-5478. doi: 10.1182/bloodadvances.2021005610. PMID: 34592758; PMCID: PMC8714709.
- Fattizzo B, Carrai V, Crugnola M, Baldacci E, Bellini M, Bosi C, Buzzatti E, Caramazza D, Carli G, Carpenedo M, Clissa C, Danesin C, De Paolis MR, Giannotta JA, Innao V, Marchetti M, Markovic U, Morotti A, Napolitano M, Patriarca A, Pettine L, Poloni A, Rivolti E, Rossi E, Santeremo TM, Santoro C, Zannier ME, Zaja F, Cantoni S, Palandri F, De Stefano V. Evans syndrome: Disease awareness and clinical management in a nation-wide ITP-NET survey. Eur J Haematol. 2024 Oct;113(4):472-476. doi: 10.1111/ejh.14256. Epub 2024 Jun 21. PMID: 39031658.
- Audia, S.; Grienay, N.; Mounier, M.; Michel, M.; Bonnotte, B. Evans’ Syndrome: From Diagnosis to Treatment. J. Clin. Med. 2020, 9, 3851. https://doi.org/10.3390/jcm9123851
- Jaime-Pérez JC, Aguilar-Calderón PE, Salazar-Cavazos L, Gómez-Almaguer D. Evans syndrome: clinical perspectives, biological insights and treatment modalities. J Blood Med. 2018 Oct 10;9:171-184. doi: 10.2147/JBM.S176144. PMID: 30349415; PMCID: PMC6190623.
- Fattizzo B, Marchetti M, Michel M, Cantoni S, Frederiksen H, Giordano G, Glenthøj A, González-López TJ, Murakhovskaya I, Napolitano M, Mingot ME, Arguello M, Patriarca A, Raso S, Vianelli N, Barcellini W. Diagnosis and management of Evans syndrome in adults: first consensus recommendations. Lancet Haematol. 2024 Aug;11(8):e617-e628. doi: 10.1016/S2352-3026(24)00144-3. Epub 2024 Jul 2. PMID: 38968944.