TL;DR
Microangiopathic hemolytic anemia or MAHA is a syndrome defined by a triad of microangiopathic damage, hemolytic anemia, and thrombocytopenia, all resulting from the physical fragmentation of red blood cells in small blood vessels.
- Pathophysiology ▾: The process is a vicious cycle where endothelial damage leads to the formation of microthrombi (tiny clots), which mechanically shear red blood cells as they pass through, causing the formation of schistocytes.
- Etiology ▾: Causes are divided into primary thrombotic microangiopathies (TMAs) like TTP and HUS, and secondary causes such as infections, malignancies, autoimmune disorders, and certain drugs.
- Clinical Presentation ▾: Symptoms include fatigue and pallor from anemia, petechiae and purpura from thrombocytopenia, and organ-specific signs like acute kidney injury (in HUS) or neurological deficits (in TTP).
- Laboratory Investigations ▾: Diagnosis relies on finding schistocytes on a peripheral blood smear, along with evidence of hemolysis (elevated LDH, decreased haptoglobin, elevated indirect bilirubin).
- Treatment ▾: Therapy is specific to the underlying cause. TTP is treated with plasma exchange, while typical HUS is managed with supportive care, and atypical HUS may require complement inhibitors.
- Prognosis ▾: The long-term outlook depends on the cause, with risks of TTP relapse and chronic kidney disease in HUS being key considerations.
- Differential Diagnosis ▾: MAHA must be distinguished from conditions like Disseminated Intravascular Coagulation (DIC), which can have similar features but is differentiated by prolonged coagulation times and significantly elevated D-dimer.
*Click ▾ for more information
Introduction
Microangiopathic hemolytic anemia or MAHA is a clinical and laboratory syndrome characterized by red blood cell fragmentation (schistocytes) and non-immune intravascular hemolysis. It is a key feature of thrombotic microangiopathies (TMAs), a group of life-threatening disorders defined by widespread microvascular thrombosis, thrombocytopenia, and organ injury.
The condition is defined by a pathological triad: microangiopathic damage (to capillaries and arterioles), hemolytic anemia, and thrombocytopenia. It is not a disease in itself, but rather a finding that points to an underlying process causing small vessel occlusion and red blood cell destruction.
Pathophysiology of MAHA and Microthrombosis
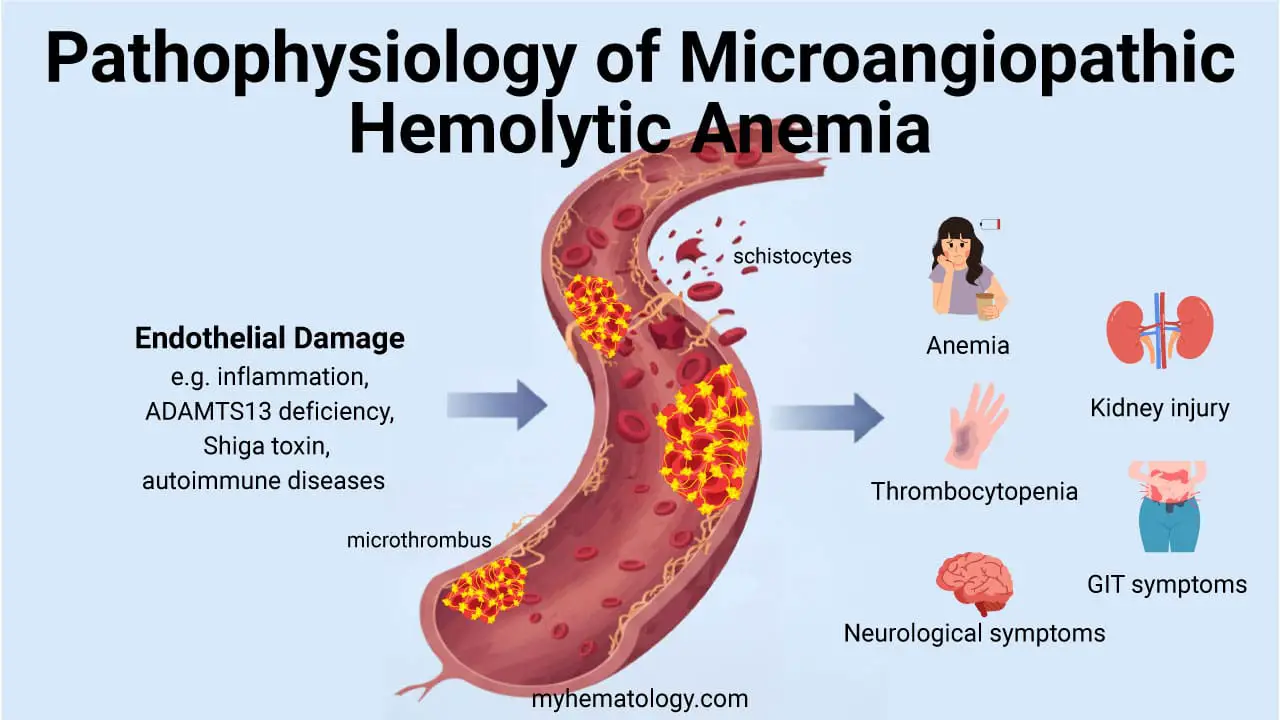
The pathophysiology of microangiopathic hemolytic anemia (MAHA) is centered on a vicious cycle of endothelial injury and microthrombi formation. The process begins with some form of damage to the inner lining of the small blood vessels (capillaries and arterioles). This damage can be triggered by a wide range of factors, such as severe infections, certain toxins, autoimmune processes, or a genetic deficiency in a key protein like ADAMTS13.
This damage to the endothelium makes the vessel surface pro-thrombotic. Platelets, which are a key component of blood clotting, are recruited to the site of injury and begin to aggregate. Fibrin, a protein involved in forming a blood clot, is also deposited. The result is the formation of small clots, or microthrombi, that partially or completely occlude the small blood vessels.
The hallmark of microangiopathic hemolytic anemia (MAHA) is the mechanical shearing of red blood cells (RBCs) as they pass through the narrowed or obstructed microvessels. The resulting sheer stress physically slices the RBCs, creating characteristic abnormal red blood cell fragments known as schistocytes on the peripheral blood smear.
As the body’s platelets and clotting factors are continuously used up to form these microthrombi, their levels in the circulating blood decrease. This explains the thrombocytopenia that is a key part of the microangiopathic hemolytic anemia (MAHA) triad. This process is usually localized to the microvasculature, which is why patients with microangiopathic hemolytic anemia (MAHA) often don’t have the widespread bleeding seen in more generalized clotting disorders like DIC.
The underlying causes of microthrombosis in microangiopathic hemolytic anemia (MAHA) are diverse, but they are broadly categorized into three main mechanisms:
- Impaired regulation of von Willebrand factor (vWF): The ultra-large vWF multimers, which are highly prothrombotic, are not properly cleaved by the enzyme ADAMTS13. This is the primary mechanism in TTP.
- Endothelial cell injury and complement dysregulation: Damage to the vascular endothelium triggers a cascade of events, including uncontrolled complement activation, which leads to platelet aggregation and microthrombi formation. This is characteristic of HUS.
- Widespread procoagulant activation: Uncontrolled activation of the coagulation cascade leads to a consumptive coagulopathy with widespread fibrin deposition and thrombin generation, as seen in DIC.
Causes of Microangiopathic Hemolytic Anemia (MAHA)
The causes of microangiopathic hemolytic anemia (MAHA) can be broadly categorized into two main groups: primary thrombotic microangiopathies (TMAs) and secondary causes.
Primary Thrombotic Microangiopathies (TMAs)
These are conditions where the central problem is the widespread formation of microthrombi, directly leading to microangiopathic hemolytic anemia (MAHA).
Thrombotic Thrombocytopenic Purpura (TTP)
The core issue in TTP is a severe deficiency in the ADAMTS13 enzyme. This enzyme’s job is to cleave large von Willebrand factor (vWF) multimers into smaller pieces. When ADAMTS13 is deficient (either due to a genetic defect or an autoimmune inhibitor), these large multimers accumulate. They become “sticky,” spontaneously binding to platelets and causing widespread, spontaneous aggregation and the formation of microthrombi in small blood vessels throughout the body.
Clinical Manifestations
The classic pentad of TTP includes:
- Fever
- Neurological symptoms (e.g., headache, confusion, seizures, stroke)
- Renal dysfunction
- MAHA
- Thrombocytopenia
However, not all five features are always present. Neurological symptoms and microangiopathic hemolytic anemia (MAHA) are the most consistent findings.
Diagnosis
Diagnosis is primarily based on clinical presentation and laboratory findings of microangiopathic hemolytic anemia (MAHA) and severe thrombocytopenia in the absence of an alternative clear cause. A key diagnostic test is the ADAMTS13 activity level. A severe deficiency (<10% activity) is highly suggestive of TTP. The PLASMIC score is a useful pre-test probability tool to guide the decision to initiate urgent therapy.
Management
TTP is a medical emergency requiring immediate treatment. The cornerstone of therapy is therapeutic plasma exchange (TPE), which removes the autoantibodies and ultra-large vWF multimers while replenishing the ADAMTS13 enzyme.
- First-line therapy: TPE and corticosteroids (e.g., prednisone, methylprednisolone).
- Adjunctive therapy: Rituximab, an anti-CD20 monoclonal antibody, is used to suppress the B-cell production of autoantibodies.
- Novel therapies: Caplacizumab, an anti-vWF nanobody, inhibits the interaction between vWF and platelets, providing a rapid clinical response and reducing the need for TPE.
Hemolytic Uremic Syndrome (HUS)
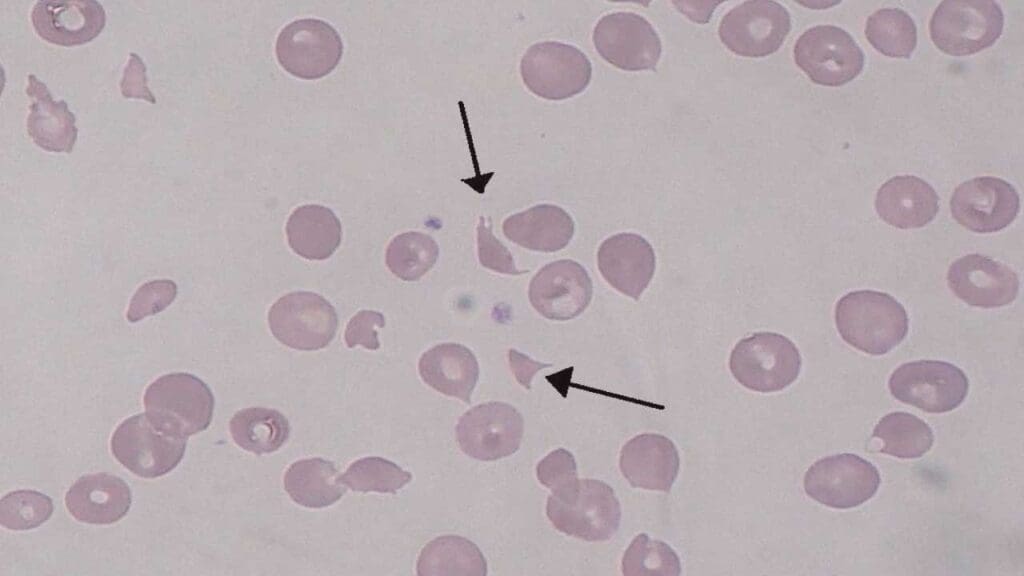
HUS is a TMA characterized by the triad of microangiopathic hemolytic anemia (MAHA), thrombocytopenia, and acute kidney injury. It is typically classified into two main subtypes: typical (STEC-HUS) and atypical (aHUS).
- Typical (Shiga Toxin-Producing E. coli HUS): The most common form of HUS is caused by an infection with certain strains of E. coli, most notably O157:H7. The bacteria produce a potent Shiga toxin that enters the bloodstream and damages the endothelium of blood vessels, especially in the kidneys. This endothelial damage triggers the formation of microthrombi and leads to the microangiopathic hemolytic anemia (MAHA) triad, with a classic presentation of acute kidney injury.
- Atypical (aHUS): This is a much rarer, genetic form of HUS. It is caused by uncontrolled activation of the complement system, a part of the innate immune system. Normally, regulatory proteins keep the complement system in check, but genetic mutations can lead to its constant, unregulated activation, which damages endothelial cells and results in microangiopathic hemolytic anemia (MAHA).
Clinical Manifestations
- STEC-HUS: Often follows a prodromal phase of bloody diarrhea and abdominal pain. The HUS symptoms (pallor, oliguria/anuria, and petechiae) typically develop 5–10 days after the onset of diarrhea.
- aHUS: Can be triggered by infections, drugs, or pregnancy, but lacks a preceding diarrheal illness. It often presents with more severe, systemic organ involvement.
Diagnosis
- STEC-HUS: Diagnosis is based on clinical presentation following bloody diarrhea and confirmation of a STEC infection via stool culture or Shiga toxin assay.
- aHUS: A diagnosis of exclusion. It is confirmed by a clinical presentation of HUS in the absence of STEC infection and often supported by genetic testing for mutations in complement-regulating genes.
Management
- STEC-HUS: Management is primarily supportive, focusing on fluid and electrolyte balance and dialysis if needed. Antibiotics are generally avoided as they may increase Shiga toxin release and worsen outcomes. Platelet transfusions are not recommended unless there is life-threatening bleeding.
- aHUS: Treatment is centered on blocking the uncontrolled complement activation. The use of complement inhibitors like eculizumab (an anti-C5 monoclonal antibody) or its successor, ravulizumab, has transformed the prognosis by preventing further endothelial damage. TPE is no longer the standard of care for aHUS.
Secondary Causes of Microangiopathic Hemolytic Anemia (MAHA)
In these cases, microangiopathic hemolytic anemia (MAHA) is a complication of another underlying condition or exposure.
- Malignancy: Certain advanced cancers, especially adenocarcinomas, can release pro-coagulant substances that activate the clotting cascade and lead to the formation of microthrombi.
- Infections: Widespread infections, particularly sepsis, can cause diffuse endothelial damage and systemic inflammation, creating an environment ripe for microthrombi formation.
- Autoimmune Disorders: Conditions like Systemic Lupus Erythematosus (SLE) or Antiphospholipid Syndrome (APS) can cause vasculitis (inflammation of blood vessels) or hypercoagulable states, leading to the small vessel damage characteristic of microangiopathic hemolytic anemia (MAHA).
- Drug-Induced: Several medications can induce microangiopathic hemolytic anemia (MAHA), often through direct endothelial toxicity. Examples include certain chemotherapy agents (e.g., mitomycin C) and immunosuppressants (e.g., cyclosporine, tacrolimus).
- Obstetric Complications: Severe conditions during pregnancy, such as pre-eclampsia and HELLP syndrome (Hemolysis, Elevated Liver enzymes, Low Platelets), are characterized by widespread endothelial dysfunction, which can trigger the microangiopathic hemolytic anemia (MAHA) process.
- Disseminated Intravascular Coagulopathy (DIC): A widespread, systemic activation of the coagulation cascade. This leads to the massive consumption of both clotting factors and platelets, resulting in both widespread microthrombi formation and a paradoxical bleeding tendency. In DIC, the coagulation times are typically prolonged, and there are very high levels of fibrin degradation products, like D-dimer.
Clinical Manifestations of MAHA
The symptoms of microangiopathic hemolytic anemia (MAHA) are a direct result of the core pathological triad highlighted: hemolytic anemia, thrombocytopenia, and organ damage caused by the microangiopathic process.
Symptoms of Anemia
The rapid destruction of red blood cells leads to a decrease in oxygen-carrying capacity. Patients will typically present with classic signs of anemia, such as profound fatigue, pallor (unusual paleness), and shortness of breath on exertion. They may also experience dizziness or a racing heart (tachycardia) as their body tries to compensate for the lack of oxygen.
Symptoms of Thrombocytopenia
The low platelet count makes it difficult for the blood to clot effectively. This can lead to various bleeding signs, including petechiae (tiny, pinpoint red or purple spots on the skin), purpura (larger areas of bruising), and spontaneous bleeding, such as nosebleeds (epistaxis) or bleeding from the gums.
Organ System Involvement
This is where the clinical picture of microangiopathic hemolytic anemia (MAHA) becomes more specific and helps distinguish between the different underlying causes. The microthrombi can form in various organs, leading to a variety of symptoms.
- Neurological Symptoms: When the microthrombi affect the small vessels in the brain, patients can experience a range of symptoms, from mild headaches and confusion to more severe signs like seizures, strokes, or altered mental status. These are particularly prominent in patients with thrombotic thrombocytopenic purpura (TTP).
- Renal Symptoms: The kidneys are often a major target for microvascular damage. Patients may develop acute kidney injury, which can manifest as reduced urine output or swelling due to fluid retention (edema). This is a hallmark feature of hemolytic uremic syndrome (HUS).
- Gastrointestinal Symptoms: In cases of typical HUS caused by a Shiga toxin-producing E. coli infection, patients often present with severe abdominal pain and bloody diarrhea before the onset of the microangiopathic hemolytic anemia (MAHA) itself.
Laboratory Investigations and Diagnosis of MAHA
The diagnosis of microangiopathic hemolytic anemia (MAHA) is rarely made on clinical grounds alone; it requires a systematic approach using laboratory tests to confirm the core triad of findings and identify the underlying cause.
Complete Blood Count (CBC) and Peripheral Blood Smear
- The Peripheral Blood Smear is the Most Critical Test: This is the cornerstone of diagnosis. The presence of schistocytes (fragmented red blood cells) is the defining pathological hallmark of microangiopathic hemolytic anemia (MAHA). These irregularly shaped fragments, often described as helmet cells, triangles, or microspherocytes, are a direct result of the mechanical shearing of red blood cells as they navigate obstructed capillaries. The quantity of schistocytes is important; a finding of more than 1% is often considered significant and highly suggestive of microangiopathic hemolytic anemia (MAHA).
- CBC Findings: The CBC will typically show anemia and thrombocytopenia (a low platelet count), which are the other two components of the microangiopathic hemolytic anemia (MAHA) triad.
Hemolysis Markers
These tests confirm that red blood cells are being destroyed within the blood vessels (intravascular hemolysis).
- Elevated Lactate Dehydrogenase (LDH): LDH is an enzyme found inside red blood cells. When these cells are destroyed, LDH is released into the bloodstream, causing a significant increase in its serum level. It’s a very sensitive marker for hemolysis.
- Elevated Indirect Bilirubin: Hemoglobin from the destroyed red blood cells is broken down into bilirubin. Since this breakdown occurs in the bloodstream and the liver has not yet processed it, the indirect (unconjugated) bilirubin level rises.
- Decreased Haptoglobin: Haptoglobin is a protein that binds to free hemoglobin in the blood to clear it from circulation. In microangiopathic hemolytic anemia (MAHA), the massive release of hemoglobin overwhelms the haptoglobin, causing its serum level to drop to very low or undetectable levels.
Coagulation Studies
This panel of tests is crucial for differentiating microangiopathic hemolytic anemia (MAHA) from other conditions, particularly Disseminated Intravascular Coagulation (DIC).
- Prothrombin Time (PT) and Activated Partial Thromboplastin Time (aPTT): In primary microangiopathic hemolytic anemia (MAHA) syndromes like TTP and HUS, the widespread microthrombi are platelet-rich but do not involve a systemic activation of the full coagulation cascade. Therefore, PT and aPTT are typically normal. This is a key distinguishing feature from DIC, where these times are often prolonged due to the consumption of clotting factors.
- D-dimer: This is a fibrin degradation product. It may be slightly elevated in microangiopathic hemolytic anemia (MAHA) due to the small, localized clots but is characteristically not as profoundly elevated as it is in DIC.
Specific Diagnostic Tests
Once MAHA is confirmed by the initial labs, these tests are used to pinpoint the specific underlying cause, as this will dictate the treatment.
- ADAMTS13 Activity: A severe deficiency (<10%) in the activity of this enzyme is diagnostic for Thrombotic Thrombocytopenic Purpura (TTP).
- Shiga Toxin Testing: A positive test for Shiga toxin in the stool or blood confirms the diagnosis of typical HUS.
- Complement Assays: Tests for complement protein levels and genetic mutations are used to diagnose atypical HUS when other causes have been ruled out.
Treatment and Management of MAHA
The treatment of microangiopathic hemolytic anemia (MAHA) is critically dependent on an accurate and timely diagnosis of the underlying cause. While general supportive care is essential for all patients, the specific therapy will vary drastically depending on whether the microangiopathic hemolytic anemia (MAHA) is due to a primary thrombotic microangiopathy (TMA) like TTP or HUS, or a secondary cause.
General Supportive Care
Regardless of the cause, several immediate measures are necessary to stabilize the patient.
- Red Blood Cell Transfusions: Patients with severe anemia may require packed red blood cell transfusions to improve oxygen delivery to tissues and alleviate symptoms like fatigue and shortness of breath.
- Platelet Transfusions (Use with Caution): Platelet transfusions are generally not recommended in primary microangiopathic hemolytic anemia (MAHA) syndromes like TTP and HUS, as they can potentially fuel the formation of more microthrombi and worsen the condition. They should be reserved only for life-threatening hemorrhage.
- Fluid and Electrolyte Management: Patients often have significant renal involvement (especially in HUS) and may require careful fluid management and correction of electrolyte imbalances.
Specific treatment based on causes for primary causes of microangiopathic hemolytic anemia (MAHA) has been discussed above. The management of secondary microangiopathic hemolytic anemia (MAHA) is focused on treating the underlying cause. For example, if the microangiopathic hemolytic anemia (MAHA) is drug-induced, the offending medication must be immediately discontinued. If it’s related to an infection, treating the infection with appropriate antibiotics is the priority. For malignancy-associated microangiopathic hemolytic anemia (MAHA), treating the cancer is the ultimate solution.
Prognosis and Long-Term Considerations
The prognosis and long-term considerations for microangiopathic hemolytic anemia (MAHA) are entirely dependent on the underlying cause.
Thrombotic Thrombocytopenic Purpura (TTP)
- Prognosis: TTP is a true medical emergency, but with modern treatment, primarily plasma exchange, the prognosis has dramatically improved. Without treatment, the mortality rate is extremely high, but with prompt and appropriate therapy, the survival rate is over 80−90%.
- Long-Term Considerations: The biggest concern is the risk of relapse. Many patients, particularly those with an autoimmune form of TTP, can have recurrent episodes. Therefore, long-term follow-up and monitoring of ADAMTS13 levels are essential. Some patients may also have long-term neurological or cognitive deficits, even after the acute episode has resolved.
Hemolytic Uremic Syndrome (HUS)
- Prognosis: The prognosis for HUS varies significantly by type.
- Typical (Shiga toxin-producing HUS): The short-term prognosis is generally good, and most children recover fully with supportive care.
- Atypical (aHUS): This form has a much poorer prognosis if left untreated, often leading to end-stage renal disease (ESRD). The introduction of complement inhibitors has dramatically improved outcomes, allowing many patients to avoid dialysis.
- Long-Term Considerations: The primary long-term concern for all forms of HUS is chronic kidney disease. Patients, especially those who required dialysis during the acute phase, are at high risk for ongoing renal dysfunction, and some may progress to ESRD, requiring long-term dialysis or a kidney transplant.
Secondary MAHA
The prognosis here is directly linked to the underlying cause. If the triggering condition can be effectively treated or removed, the microangiopathic hemolytic anemia (MAHA) will usually resolve, and the prognosis is good. For example, if the microangiopathic hemolytic anemia (MAHA)is drug-induced, simply stopping the medication can lead to a full recovery. However, if the microangiopathic hemolytic anemia (MAHA) is a complication of a serious, ongoing condition like widespread cancer or severe sepsis, the prognosis is often poor, reflecting the severity of the primary disease.
Differential Diagnosis of MAHA
The differential diagnosis for MAHA primarily involves other causes of anemia and thrombocytopenia. The most crucial distinction to make is with Disseminated Intravascular Coagulation (DIC), but other conditions should also be considered.
| Feature | Microangiopathic Hemolytic Anemia (MAHA) | Disseminated Intravascular Coagulation (DIC) |
| Pathophysiology | Mechanical shearing of red blood cells. | Systemic activation of the coagulation cascade. |
| Peripheral Smear | Schistocytes present and abundant. | Schistocytes may be present but less prominent. |
| Thrombocytopenia | Present. | Present and often severe. |
| Anemia | Hemolytic anemia due to red cell fragmentation. | Anemia due to red cell fragmentation and bleeding. |
| PT & aPTT | Normal | Prolonged |
| D-dimer | Mildly elevated. | Significantly elevated. |
| Key Distinction | Schistocytes with normal coagulation studies. | Prolonged PT/aPTT with consumptive coagulopathy. |
Conclusion
Microangiopathic hemolytic anemia is a critical finding that demands prompt investigation and differentiation of its underlying cause. While sharing a common feature of RBC fragmentation, the distinct pathophysiological mechanisms of TTP, HUS, and DIC necessitate a targeted diagnostic approach and specific, often urgent, therapeutic interventions.
Frequently Asked Questions (FAQs)
What is the most important laboratory finding for microangiopathic hemolytic anemia (MAHA)?
The most important finding is the presence of schistocytes on the peripheral blood smear. These fragmented red blood cells are a direct visual confirmation of the underlying mechanical hemolysis.
How to differentiate between TTP and HUS?
While both are forms of TMA, the key differentiator is the underlying cause and severity of certain symptoms. TTP is characterized by a severe ADAMTS13 deficiency and prominent neurological symptoms. HUS, particularly STEC-HUS, is often preceded by a diarrheal illness and is primarily associated with severe acute kidney injury.
Can microangiopathic hemolytic anemia (MAHA) be a sign of cancer?
Yes, MAHA can be a paraneoplastic syndrome, particularly in advanced adenocarcinomas (e.g., gastric, lung, breast cancer). It is thought to be caused by procoagulant substances released by the tumor, leading to a form of chronic DIC.
Is it ever safe to give platelet transfusions in a patient with TTP?
Platelet transfusions are generally contraindicated in TTP unless there is life-threatening hemorrhage. Giving platelets can fuel the formation of microthrombi and worsen the clinical picture. The primary goal is to address the underlying cause (ADAMTS13 deficiency) with plasma exchange.
What is the difference between microangiopathic and macroangiopathic hemolytic anemia?
Microangiopathic hemolytic anemia (MAHA) and macroangiopathic hemolytic anemia are distinguished by the location of red blood cell destruction. In MAHA, the fragmentation of red blood cells occurs in small blood vessels (capillaries and arterioles), typically due to mechanical shearing as the cells pass through a microvasculature obstructed by platelet-fibrin clots, a hallmark of conditions like TTP and HUS.
Conversely, in macroangiopathic hemolytic anemia, red blood cell destruction occurs in large blood vessels or the heart, caused by physical damage from turbulent blood flow or the presence of a prosthetic device, such as a mechanical heart valve. The core difference, therefore, lies in the scale of the pathological process, occurring at the microvascular versus the macrovascular level.
Glossary of Medical Terms
- ADAMTS13: A disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13. It is an enzyme that cleaves von Willebrand factor (vWF) multimers. A deficiency leads to TTP.
- Consumptive Coagulopathy: A condition in which the proteins and platelets involved in blood clotting are used up faster than they can be produced, leading to widespread clotting and bleeding.
- Endothelial Cells: The cells that line the interior surface of blood vessels and lymphatic vessels.
- Hemolysis: The destruction or breakdown of red blood cells.
- Microthrombosis: The formation of tiny blood clots within the smallest blood vessels.
- Peripheral Blood Smear: A laboratory test in which a drop of blood is smeared on a glass slide, stained, and examined under a microscope. It is crucial for identifying schistocytes.
- Schistocytes: Fragmented red blood cells, a key indicator of mechanical hemolysis and MAHA.
- Thrombocytopenia: A condition characterized by an abnormally low number of platelets in the blood.
- Von Willebrand Factor (vWF): A blood glycoprotein involved in hemostasis, which plays a role in platelet adhesion to the vessel wall at the site of a wound.
Disclaimer: This article is intended for informational purposes only and is specifically targeted towards medical students. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Pande, A., Kumar, A., Krishnani, H., Acharya, S., & Shukla, S. (2023). Recent Advances in the Management of Microangiopathic Hemolytic Anemias (MAHA): A Narrative Review. Cureus, 15(10), e47196. https://doi.org/10.7759/cureus.47196
- Sohail, M. A., Luong, P., Sedor, J., & Kaw, R. (2022). Microangiopathic hemolytic anemia in a female patient with systemic lupus erythematosus. Cleveland Clinic journal of medicine, 89(3), 130–138. https://doi.org/10.3949/ccjm.89a.21066
- Thomas, M. R., & Scully, M. (2021). How I treat microangiopathic hemolytic anemia in patients with cancer. Blood, 137(10), 1310–1317. https://doi.org/10.1182/blood.2019003810.
- Lechner, Klaus MD; Obermeier, Hanna Lena. Cancer-Related Microangiopathic Hemolytic Anemia: Clinical and Laboratory Features in 168 Reported Cases. Medicine 91(4):p 195-205, July 2012. https://doi.org/10.1097/MD.0b013e3182603598