TL;DR
Platelet or a thrombocyte is a small, anucleated cellular fragment derived from bone marrow megakaryocyte.
- Lifespan & Count: They circulate for 7–10 days with a normal reference range of 150,000 – 450,000/µL.
- Morphology ▾:
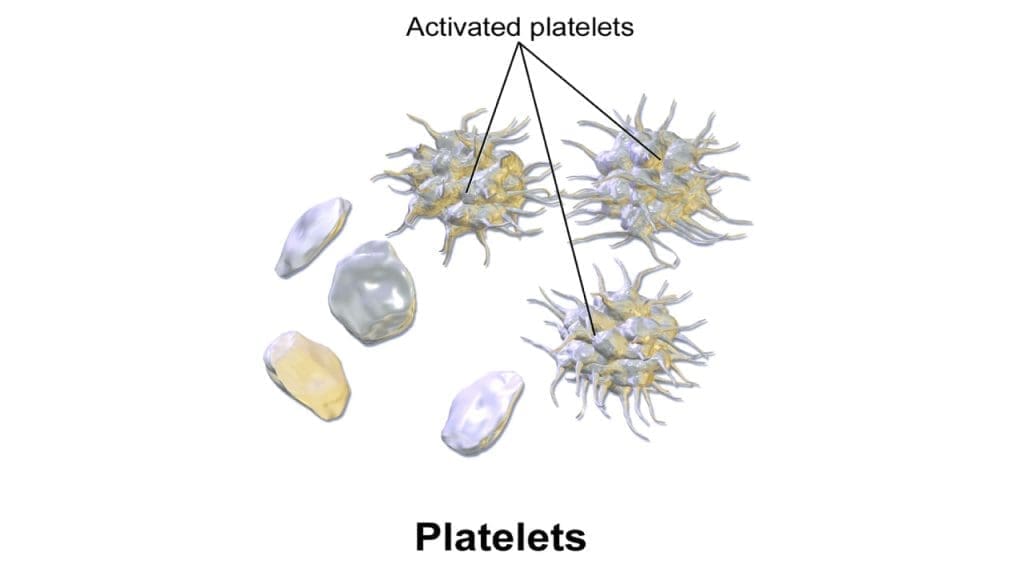
- Resting: Smooth, discoid shape to flow easily.
- Activated: Transforms into spiky spheres with pseudopods to grab surfaces.
- Primary Function (Hemostasis):
- Adhesion: Stick to exposed collagen via von Willebrand Factor (vWF).
- Activation: Release granules (ADP, Thromboxane A2) to call for help.
- Aggregation: Bind to each other using Fibrinogen bridges to form a “platelet plug.”
- Key Regulation:
- Thrombopoietin (TPO): The main hormone (from the liver) that stimulates platelet production.
- Inhibitors: Healthy blood vessels secrete Nitric Oxide and Prostacyclin to prevent platelets from clotting unnecessarily.
- Disorders:
- Thrombocytosis (High) ▾: Can be Primary (Essential Thrombocythemia) or Secondary (due to inflammation/infection). Risk of thrombosis.
- Thrombocytopenia (Low) ▾: caused by Decreased Production (bone marrow failure), Increased Destruction (ITP), or Sequestration (enlarged spleen). Risk of bleeding.
- Clinical Signs ▾: Low platelets typically present with mucosal bleeding (gums, nose) and petechiae (pinpoint skin spots).
- Key Labs ▾: CBC (count), Peripheral Smear (morphology), and Platelet Function Tests (aggregation studies).
*Click ▾ for more information
Introduction
Platelets are tiny cellular fragments that play a crucial role in the process of blood clotting, known as hemostasis. They are produced in the bone marrow from larger cells called megakaryocytes.
When a vessel wall is breached, platelets are the first responders. Drawn by the siren signal of exposed collagen, they undergo a dramatic shape change – transforming from placid discs to spiky spheres. Armed with adhesive glycoproteins like GPIb-IX and αIIbβ3 integrins, they firmly grip the collagen matrix, forming the initial plug.
But thrombocytes are not solitary warriors. They activate a cascade of signaling, releasing potent agonists like adenosine diphosphate (ADP) and thromboxane A2. These molecules, like potent battle cries, summon additional platelets to the scene. Through fibrinogen bridges, platelets form an intricate web, a living barricade against further hemorrhage.
Unchecked platelet activation and aggregation can culminate in the formation of unwanted clots and thrombosis within the vasculature.
Understanding the intricate interplay of G-protein coupled receptors, calcium signaling, and cytoskeletal rearrangements is crucial. This knowledge will equip us to delve deeper into the mysteries of hemostasis and thrombosis, paving the way for future advancements in therapeutic interventions.
A Historical Journey Through Platelet Discovery
Early Observations (18th-19th Centuries)
- 1733: Giovanni Battista Morgagni: While dissecting a patient who died from hemorrhage, Morgagni observed tiny white bodies in the blood. He attributed them to “milky particles” but lacked the technology to understand their function.
- 1842: William Addison: Addison, a physician, described “clusters of corpuscles” in the blood of patients with purpura, suggesting their possible role in blood clotting.
- 1882: Giulio Bizzozero: The Italian physician Bizzozero is often credited with “discovering” platelets. He observed them in live frog blood, noting their distinct morphology and their role in forming white thrombi (clots). He named them “piastrine,” which translates to “little plates” in Italian.
Expanding the Knowledge (20th Century and Beyond)
- 1906-1910: James Homer Wright: This American pathologist further characterized platelets, demonstrating their role in blood clotting through experiments with rabbits. He also coined the term “thrombocytes,” highlighting their involvement in clot formation.
- 1930s-1940s: Studies on Platelet Function: Researchers like Karl Landsteiner and Max Bergmann began investigating the biochemical mechanisms of platelet aggregation and clot formation, identifying key players like fibrinogen and ADP.
- 1960s-Present: Advances in electron microscopy and molecular biology revealed the intricate structure and signaling pathways within platelets, leading to a deeper understanding of their activation and function. The discovery of specific receptors, like GPIb-IX and integrins, paved the way for the development of targeted antiplatelet therapies.
Key Milestones and Figures
- Development of the first electron microscopes: This technology allowed scientists to visualize the detailed structure of thrombocytes, including their internal granules and cytoskeletal elements.
- Discovery of von Willebrand factor (vWF): This protein plays a crucial role in platelet adhesion to the injured vessel wall, providing a crucial link in the hemostatic cascade.
- Identification of platelet-specific receptors and signaling pathways: Understanding these pathways opened doors for the development of targeted therapies for bleeding and thrombotic disorders.
Platelet Morphology and Ultrastructure
Unlike their spherical counterparts, the red and white blood cells, platelets boast a unique discoid shape. This flattened structure, with a central bulge and thin margins, offers several functional advantages:
- Enhanced Area-to-Volume Ratio: The large surface area provided by the discoid form facilitates rapid interaction with the vessel wall and other blood components during clotting. This allows for efficient adhesion to exposed collagen and the formation of a strong, tight plug.
- Increased Membrane Fluidity: The thin margins enable rapid membrane bending and deformation, crucial for shape changes during activation. This fluidity allows platelets to readily transform from smooth discs into spiky spheres, maximizing their adhesive capabilities.
- Enhanced Flow Dynamics: The discoid shape minimizes friction with surrounding blood cells, aiding in smooth flow through the vasculature. This prevents unnecessary activation and aggregation, maintaining the delicate balance between hemostasis and normal blood flow.
From Rest to Activation
While resting in the bloodstream, platelets exhibit a smooth discoid shape, minimizing their interaction with other components. However, upon encountering a site of vascular injury, they undergo a dramatic transformation. This activation triggers a cascade of events, leading to significant changes in shape:
- Marginal Protrusions: The thin margins begin to develop spikes and pseudopods, increasing their surface area and facilitating adhesion to collagen and other thrombocytes.
- Central Contraction: The central bulge contracts, further enhancing the discoid shape and promoting tighter packing of platelets within the forming clot.
- Spherical Transformation: In some cases, platelets may even completely transform into spiky spheres, maximizing their adhesive potential and contributing to a stronger clot structure.
Functional Implications
These shape changes are not merely aesthetic; they have profound functional consequences:
- Enhanced Adhesion: The increased surface area and protrusions facilitate stronger binding to collagen and vWF, anchoring the clot to the injured vessel wall.
- Intercellular Communication: The closer packing of activated platelets allows for efficient propagation of signaling molecules like ADP and thromboxane A2, promoting further activation and aggregation.
- Clot Stabilization: The spiky spheres interlock and form a tight mesh, reinforcing the clot structure and preventing bleeding.
Membrane Composition
The thrombocyte membrane is composed of three crucial elements: the phospholipid bilayer, the glycocalyx, and a diverse array of integral membrane proteins – each playing a vital role in the platelet’s function.
Phospholipid Bilayer
The fluid phospholipid bilayer allows for:
- Rapid Shape Changes: The fluidity of the bilayer enables platelets to readily transform their discoid shape into spiky spheres during activation, maximizing their surface area for adhesion.
- Lateral Movement of Proteins: Integral membrane proteins like integrins and selectins can move freely within the bilayer, positioning themselves strategically for efficient interaction with other cells and molecules.
- Maintenance of Membrane Integrity: The bilayer acts as a barrier, protecting the platelet’s internal machinery from external factors and maintaining its internal environment.
Glycocalyx
Adorning the outer surface of the phospholipid bilayer, the glycocalyx plays several crucial roles:
- Initial Contact and Rolling: The glycocalyx facilitates initial contact with the vessel wall by interacting with negatively charged molecules like von Willebrand factor (vWF). This interaction allows platelets to roll along the endothelium, searching for sites of injury.
- Negative Charge Barrier: The dense network of sugar molecules creates a negatively charged barrier, preventing unwanted adhesion to healthy endothelial cells and maintaining normal blood flow.
- Signaling Platform: The glycocalyx harbors specific sugar sequences that serve as docking sites for signaling molecules like P-selectin, initiating important activation pathways.
Integral Membrane Proteins
Embedded within the phospholipid bilayer are a diverse array of integral membrane proteins, each with a specific role in adhesion and signaling.
- Integrins: These versatile adhesion molecules act like molecular handshakes, connecting platelets to collagen, vWF, and other platelets through specific ligand binding. Different integrin subtypes are activated during different stages of clotting, ensuring a coordinated and efficient response.
- Selectins: These sugar-binding proteins guide platelets towards sites of injury by interacting with the glycocalyx of endothelial cells. P-selectin on the endothelium, for example, captures rolling platelets, bringing them closer to the site of damage.
- Receptors: Various receptors, like G-protein coupled receptors, sense specific molecules like ADP and thromboxane A2, triggering downstream signaling pathways for platelet activation and aggregation.
- Channels: Ion channels like Ca2+ channels regulate the influx of calcium ions, a crucial second messenger for platelet activation and shape change.
Adhesion and Signaling
These integral membrane proteins do not work in isolation. They collaborate and orchestrate a complex dance of adhesion and signaling:
- Integrin Activation: Selectins initially tether platelets, bringing them close to the injured vessel wall. Integrins, initially inactive, are then activated by specific signals, allowing them to bind firmly to collagen and fibrinogen, forming the initial clot.
- Signaling Cascades: Receptors like G-protein coupled receptors initiate intracellular signaling cascades, leading to shape changes, granule release, and further activation of integrins and other signaling molecules.
- Clot Consolidation: Activated integrins on adjacent platelets bind to fibrinogen, bridging them together and solidifying the clot structure.
Cytoskeleton
The platelet cytoskeleton is a dynamic network of microtubules, microfilaments, and an actin-based gel-sol system, crucial for maintaining the discoid shape and orchestrating the diverse functions of these microscopic guardians of hemostasis.
Microtubules
These hollow tubes, composed of α-tubulin and β-tubulin subunits, act as the internal scaffolding of the platelet. They run parallel to the long axis of the discoid shape, providing a framework for maintaining its integrity and stability during rest and activation. Microtubules also play a vital role in:
- Organelle Positioning: They guide the movement and positioning of internal organelles, ensuring efficient granule release and signaling during activation.
- Shape Maintenance: They resist deformation forces, preventing the platelet from becoming overly spherical during activation, thereby enhancing its adhesive capabilities.
- Movement and Transport: Microtubules serve as tracks for motor proteins, facilitating the transport of vesicles and other components within the platelet.
Microfilaments
These thin, solid filaments, composed of actin and myosin, are the workhorses of the platelet cytoskeleton. They are responsible for the dynamic shape changes that occur during activation, including:
- Marginal Protrusions: During activation, actin filaments polymerize rapidly, forming spikes and pseudopods on the platelet margins, increasing surface area for adhesion and intercellular communication.
- Central Contraction: Myosin motor proteins cross-link and pull actin filaments, causing the central bulge of the platelet to contract, further enhancing its adhesive potential.
- Clot Retraction: After forming a clot, actin and myosin work together to contract the clot, pulling the vessel walls closer together and promoting wound healing.
The Actin-Based Gel-Sol System
This remarkable system is a semi-fluid network of actin filaments that constantly transitions between a gel-like state, providing rigidity and shape, and a sol-like state, enabling rapid remodeling. This dynamic dance of polymerization and depolymerization allows the platelet to adapt its internal structure to different functional demands:
- Resting State: The gel state predominates, maintaining the discoid shape and preventing unnecessary activation.
- Activation State: The sol state increases, allowing for rapid actin filament rearrangement and shape changes crucial for adhesion and aggregation.
- Clot Consolidation: The gel state reasserts itself, stabilizing the clot structure and promoting wound healing.
These different components of the platelet cytoskeleton work together in a coordinated manner for movement and function:
- Microtubules provide the stable framework, while microfilaments and the actin-based gel-sol system execute the dynamic movements.
- Signaling pathways triggered during activation regulate the polymerization and depolymerization of actin filaments, directing the specific shape changes required for each function.
- The interplay between these components allows the platelet to adapt its shape and function seamlessly, ensuring efficient hemostasis and maintaining vascular integrity.
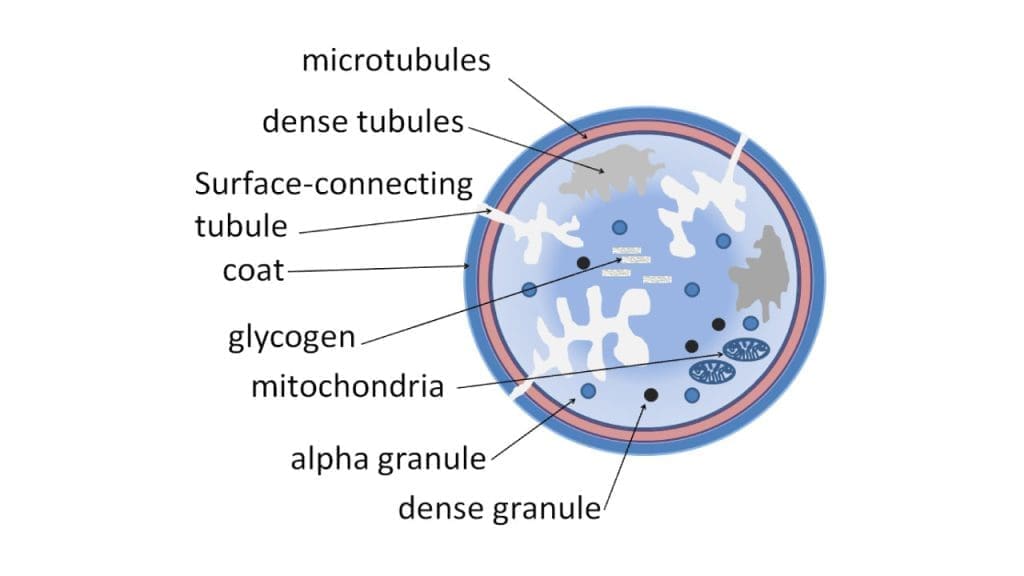
Platelet Storage Granules
Platelet storage granules are specialized organelles packed with potent molecules that are the secret weapons in the platelet’s hemostatic arsenal.
Alpha Granules
These larger granules, constituting about 70% of the total platelet granule pool, are packed with a diverse array of molecules that serve various functions:
- Coagulation Factors: These proteins, like fibrinogen, vWF, and factor V, are crucial for the formation and stabilization of the fibrin clot, the final barrier against bleeding.
- Adhesion Molecules: Von Willebrand factor (vWF) and fibronectin not only contribute to clot formation but also play a critical role in anchoring platelets to the injured vessel wall through specific interactions with collagen and endothelial cells.
- Chemotactic and Proliferative Factors: Platelet-derived growth factor and other growth factors promote the migration and proliferation of endothelial cells and smooth muscle cells, facilitating wound healing and vascular repair.
- Serotonin: This potent vasoconstrictor helps narrow blood vessels at the site of injury, minimizing further blood loss.
- Antithrombin III: This natural anticoagulant regulates the coagulation cascade, preventing unwanted clot formation beyond the immediate area of injury.
Dense Granules
These smaller, denser granules, constituting about 30% of the pool, are packed with molecules that drive platelet aggregation and clot stabilization:
- ADP (Adenosine Diphosphate): This potent platelet activator triggers further shape change, granule release, and aggregation in a cascading effect.
- Thromboxane A2: This lipid mediator promotes platelet aggregation and vasoconstriction, reinforcing the clot and minimizing blood loss.
- Calcium: This crucial second messenger regulates various signaling pathways and functions within the platelet, including granule release and cytoskeletal rearrangements.
Release Mechanisms
Upon encountering a site of injury, platelets undergo a rapid activation process that triggers the release of these potent molecules from their respective granules:
- Exocytosis: This regulated mechanism utilizes SNARE complexes to fuse the granule membranes with the plasma membrane, releasing the contents into the extracellular space.
- Secondary Activation: Released ADP binds to purinergic receptors on neighboring platelets, triggering their activation and further granule release, amplifying the hemostatic response.
- Calcium Signaling: Calcium influx during activation triggers various signaling pathways, including the release of granule contents through exocytosis.
Mitochondria and Endoplasmic Reticulum
Though often considered nuclei-less remnants, platelets boast a surprising degree of metabolic and synthetic prowess, thanks to the presence of two key organelles: mitochondria and the endoplasmic reticulum (ER).
Mitochondria
Despite their small size, platelets pack 5-8 mitochondria, miniature powerhouses that generate the ATP (adenosine triphosphate) needed for various functions:
- Shape Change and Movement: Rapidly remodeling their cytoskeleton during activation requires significant energy, which mitochondria provide through oxidative phosphorylation.
- Granule Release: Exocytosis of stored hemostatic molecules like ADP and serotonin is an energy-intensive process fueled by ATP.
- Calcium Signaling: Maintaining calcium homeostasis, crucial for platelet activation and function, relies on ATP-powered pumps in the mitochondrial membrane.
However, platelet mitochondria aren’t just energy factories. They also participate in:
- Reactive Oxygen Species (ROS) Production: Low levels of ROS act as signaling molecules, but excessive production can damage platelets and contribute to thrombosis.
- Apoptosis: When platelets are damaged or aged, their mitochondria trigger programmed cell death, preventing rogue platelets from causing harm.
Endoplasmic Reticulum
The ER, a complex network of membranes, is the platelet’s protein synthesis hub. It produces and folds various proteins critical for:
- Membrane Repair: Platelets constantly experience stress, and the ER supplies proteins for repairing their plasma membrane.
- Granule Content: Fibrinogen, vWF, and other hemostatic molecules stored in granules are synthesized and processed in the ER.
- Platelet Activation Receptors: The ER manufactures and transports receptors like GPIb-IX and selectins to the platelet surface, enabling them to sense and respond to injury signals.
Beyond protein synthesis, the ER also plays a role in
- Calcium Storage: The ER acts as a reservoir for calcium ions, releasing them during activation to trigger downstream signaling events.
- Stress Response: When platelets encounter stress, the ER initiates an unfolded protein response to maintain protein quality and prevent dysfunction.
The Impact on Platelet Function
Disruptions in mitochondrial or ER function can significantly impact platelet function:
- Mitochondrial Dysfunction: Reduced ATP production can lead to impaired shape change, granule release, and calcium signaling, hindering effective clot formation.
- ER Stress: Excessive protein misfolding can trigger apoptosis, leading to premature platelet death and a reduced lifespan.
- Thrombosis: Mitochondrial ROS production and ER-mediated platelet activation can contribute to unwanted clot formation in certain conditions.
Platelet Activation and Signaling Pathways
There are diverse triggers that lead to platelet activation.
Vessel Wall Injury
The primary trigger for platelet activation is the injured vessel wall. When endothelial cells lining the blood vessel are damaged, the underlying collagen, a potent platelet activator, is exposed. This acts like a distress signal, initiating a cascade of events:
- GPIb-IX Recognition: Collagen binds to the GPIb-IX receptor on the platelet surface, triggering initial tethering and slowing the platelet’s flow.
- Signal Transduction: Intracellular signaling pathways are activated, leading to cytoskeletal rearrangements, shape change, and the release of granule contents like ADP.
- Amplification and Aggregation: Released ADP binds to purinergic receptors on other platelets, activating them and further amplifying the response. This cascade leads to the formation of a platelet plug, sealing the injured site and preventing blood loss.
ADP Release
ADP, released from activated platelets and damaged endothelial cells, acts as a potent secondary activator, amplifying the initial response:
- Purinergic Receptor Activation: ADP binds to P2Y12 receptors on the platelet surface, triggering intracellular signaling pathways.
- Calcium Mobilization: Calcium ions flood the cytosol, acting as second messengers and activating downstream events like granule release and integrin activation.
- Shape Change and Aggregation: Increased calcium promotes cytoskeletal rearrangements and the activation of integrins like αIIbβ3, which bind fibrinogen and bridge adjacent platelets, forming a stable clot.
Thrombin Generation
The coagulation cascade, triggered by the injured vessel wall, culminates in the formation of thrombin, another potent platelet activator:
- Protease-Activated Receptors (PARs): Thrombin activates PARs on the platelet surface, triggering similar signaling pathways as ADP, leading to calcium mobilization, shape change, granule release, and integrin activation.
- Clot Stabilization: Thrombin also cleaves fibrinogen into fibrin, which polymerizes and strengthens the existing platelet plug, further preventing blood loss.
Other Stimuli
Beyond these major triggers, various other stimuli can contribute to platelet activation:
- Immune Complexes: Complexes of antibodies and antigens can activate platelets through Fc receptors.
- Endothelial Signals: Damaged endothelial cells can release factors like thrombomodulin, which activates PARs on platelets.
- Bacterial and Viral Products: Certain pathogens can activate platelets directly or indirectly through inflammatory pathways.
G-protein coupled receptors (GPCRs) in platelets
GPCRs are membrane-bound proteins that sense specific ligands, like ADP or thrombin, and initiate a cascade of intracellular signaling events. They don’t work alone; they partner with G-proteins, heterotrimeric proteins composed of alpha, beta, and gamma subunits. Upon ligand binding, the GPCR activates the G-protein, triggering the dissociation of the alpha subunit from the beta-gamma complex.
G-α
The freed alpha subunit activates various downstream effector molecules, including:
- Phospholipase C (PLC): PLC cleaves phosphatidylinositol 4,5-bisphosphate (PIP2) into inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG).
- Protein Kinase C (PKC): DAG activates PKC, which phosphorylates various target proteins, influencing platelet function.
- Calcium Channels: IP3 mobilizes calcium ions from intracellular stores, further amplifying the signaling cascade.
- Rho GTPases: These GTP-binding proteins regulate cytoskeletal rearrangements, crucial for shape changes.
Shape Change, Granule Release, and Aggregation
These activated effector molecules orchestrate the key downstream effects of GPCR signaling:
- Shape Change: Rho GTPases, activated by G-α, promote the formation of actin filaments and pseudopods, causing the platelet to transform from a discoid shape into a spiky sphere, maximizing its adhesive potential.
- Granule Release: Calcium mobilization, triggered by IP3, and PKC activation lead to the phosphorylation of proteins involved in granule fusion with the plasma membrane, releasing potent hemostatic molecules like ADP, serotonin, and coagulation factors.
- Aggregation: Released ADP activates other GPCRs on neighboring platelets, propagating the signal and leading to their activation and aggregation through integrin-mediated bridging with fibrinogen.
Specific GPCRs and Their Effects
Platelets express various GPCRs, each with unique roles:
- P2Y12 receptors: Activated by ADP, they contribute primarily to shape change and granule release.
- Thromboxane A2 receptors: Activated by thromboxane A2, they promote aggregation and vasoconstriction.
- Protease-activated receptors (PARs): Activated by thrombin, they trigger diverse effects, including shape change, granule release, and aggregation.
- Serotonin receptors: Activated by released serotonin, they contribute to vasoconstriction and platelet activation.
Platelet Calcium Signaling
Platelet activation is triggered by various stimuli like collagen exposure and ADP release, leading to a surge in calcium ions.
- Calcium Influx: Specific channels in the plasma membrane, activated by G-protein coupled receptors and other signaling pathways, allow calcium ions to flood into the cytosol.
- Intracellular Mobilization: Calcium is also released from internal stores like the endoplasmic reticulum, further amplifying the intracellular concentration.
Downstream Effects of Calcium
This elevated intracellular calcium acts as a second messenger, triggering a cascade of downstream events.
- Shape Change: Calcium activates Rho GTPases, proteins that regulate the cytoskeleton. This leads to the formation of actin filaments and pseudopods, transforming the platelet from a smooth disc to a spiky sphere, maximizing its adhesive surface area.
- Granule Release: Calcium binds to proteins involved in granule fusion, causing the release of potent hemostatic molecules like ADP, serotonin, and coagulation factors. These factors contribute to clot formation and vasoconstriction, minimizing blood loss.
- Integrin Activation: Calcium promotes the activation and clustering of integrins, especially αIIbβ3, which bind to fibrinogen and bridge adjacent platelets, forming a stable clot structure.
- Signaling Amplification: Released ADP binds to purinergic receptors on other platelets, triggering their activation and further amplifying the calcium signaling cascade. This positive feedback loop ensures a rapid and efficient hemostatic response.
Maintaining Calcium Homeostasis
Excessive or prolonged elevation of intracellular calcium can lead to detrimental effects.
- Platelet Dysfunction: Excessive calcium can inhibit further activation and impair clot retraction, hindering effective hemostasis.
- Apoptosis: High calcium levels can trigger programmed cell death, prematurely eliminating activated platelets.
- Thrombosis: Uncontrolled activation can lead to the formation of unwanted clots, potentially obstructing blood flow.
Therefore, platelets utilize various mechanisms to maintain calcium homeostasis.
- Calcium Pumps: These proteins actively transport calcium ions back into the endoplasmic reticulum or out of the cell.
- Calcium Binding Proteins: These proteins sequester calcium within the cytosol, preventing its uncontrolled activation of downstream pathways.
Thromboxane A2
Thromboxane A2 (TXA2) is a key player in the drama of platelet activation and clot formation
TXA2 Synthesis
When platelets encounter stimuli like collagen exposure or ADP release, they activate a cascade of events leading to TXA2 synthesis.
- Phospholipase A2: This enzyme cleaves arachidonic acid from phospholipids in the platelet membrane, creating the fuel for TXA2 synthesis.
- Thromboxane Synthase: This enzyme transforms arachidonic acid into TXA2, the potent final product.
Effects on Platelet Function
Once synthesized, TXA2 rapidly exerts its influence on platelet behavior through specific G-protein coupled receptors (GPCRs).
- Platelet Aggregation: TXA2 binds to thromboxane A2 receptors (TP receptors) on the platelet surface, activating G-proteins and triggering downstream signaling pathways. This leads to cytoskeletal rearrangements and integrin activation, causing platelets to aggregate and form a stable clot.
- Shape Change: TXA2 signaling also contributes to platelet shape change, promoting the formation of pseudopods that increase the surface area for adhesion and aggregation.
- Vasoconstriction: TXA2 acts as a potent vasoconstrictor, stimulating smooth muscle cells in the blood vessel wall to contract. This narrows the vessel diameter, minimizing blood loss and further promoting clot formation.
The Delicate Balance
While TXA2 plays a crucial role in hemostasis, its potent effects need to be tightly regulated to prevent unwanted consequences.
- Thrombosis: Uncontrolled TXA2 production can lead to excessive platelet aggregation and clot formation, potentially obstructing blood flow and causing ischemic events.
- Vascular Damage: TXA2-induced vasoconstriction can restrict blood flow and damage tissues if not controlled.
Therefore, mechanisms exist to limit TXA2 activity:
- Rapid Degradation: TXA2 has a short half-life, quickly breaking down into inactive metabolites to prevent prolonged activation.
- Competitive Inhibition: Aspirin irreversibly inhibits thromboxane synthase, preventing TXA2 synthesis and reducing platelet aggregation.
Phosphoinositide 3-Kinase (PI3K) Pathway
Phosphoinositide 3-kinase (PI3K) pathway is a complex series of signaling molecules that plays a crucial role in orchestrating platelet function.
The PI3K pathway involves a cascade of proteins:
- PI3K: This enzyme phosphorylates phosphatidylinositol lipids, creating signaling molecules that activate downstream pathways.
- Akt: This protein kinase, once activated by PI3K, triggers various cellular processes.
- Additional Signaling Molecules: Other proteins like Ras GTPases and phosphoinositide phosphates orchestrate the flow of information within the pathway.
PI3K Activation in Platelets
Various stimuli, like collagen exposure and ADP release, can activate the PI3K pathway in platelets.
- Receptor Activation: G-protein coupled receptors (GPCRs) and integrins on the platelet surface sense specific ligands and trigger intracellular signaling cascades.
- PI3K Recruitment: These pathways activate and recruit PI3K to the plasma membrane.
Downstream Effects of PI3K
Once activated, PI3K phosphorylates phosphatidylinositol lipids, leading to a cascade of downstream effects:
- Shape Change: Activated Akt promotes cytoskeletal rearrangements, transforming the platelet from discoid to spiky, maximizing its adhesive surface area.
- Granule Release: Akt signaling triggers granule fusion with the plasma membrane, releasing potent hemostatic molecules like ADP, serotonin, and coagulation factors.
- Integrin Activation: Akt also promotes the activation and clustering of integrins, particularly αIIbβ3, which bind fibrinogen and bridge platelets, stabilizing the clot.
- Positive Feedback Loop: Released ADP activates other platelets’ GPCRs, further propagating the PI3K signaling cascade and amplifying the hemostatic response.
PI3K Isoforms in Platelets
Platelets express three distinct PI3K isoforms:
- PI3Kβ: This isoform plays a primary role in ADP-induced activation, affecting shape change and granule release.
- PI3Kγ: This isoform participates in collagen-induced activation, influencing integrin activation and clot formation.
- PI3Kα: This isoform appears to have a less prominent role in platelet function, though its specific contributions are still being investigated.
Targeting the PI3K Pathway
The PI3K pathway’s crucial role in platelet activation makes it a potential target for therapeutic interventions in:
- Thrombosis: Inhibiting specific PI3K isoforms could offer a targeted approach to preventing excessive platelet aggregation and clot formation in thrombotic disorders.
- Bleeding Disorders: Understanding how PI3K interacts with other pathways could lead to the development of novel therapies to promote platelet function and address bleeding diatheses.
Platelet Adhesion and Aggregation
Adhesion to Exposed Subendothelial Matrix
Key Adhesive Glycoproteins
- GPIb-IX: This receptor complex acts as the first responder, a sticky hand that grabs onto vWF tethered to the exposed collagen or fibronectin. It slows down the platelet and initiates the adhesion process.
- Integrins: These multi-legged giants, especially αIIbβ3, come into play later, acting as the main anchors. They firmly bind to exposed collagen fibrils and fibronectin, solidifying the adhesion and laying the foundation for clot formation.
Specific Interactions
- Collagen: The main attraction in the subendothelial matrix, collagen binds to both GPIb-IX and integrins. GPIb-IX’s initial tethering gives time for integrins to unfold their legs and latch onto collagen with a strong, specific grip.
- Fibronectin: A versatile player, fibronectin can bind to both GPIb-IX and integrins. It acts as a backup, supporting and strengthening adhesion when collagen is scarce.
- Von Willebrand Factor (vWF): vWF bridges the gap between platelets and the subendothelial matrix. It binds to GPIb-IX on the platelet surface and collagen or fibronectin in the tissue, forming a crucial link.
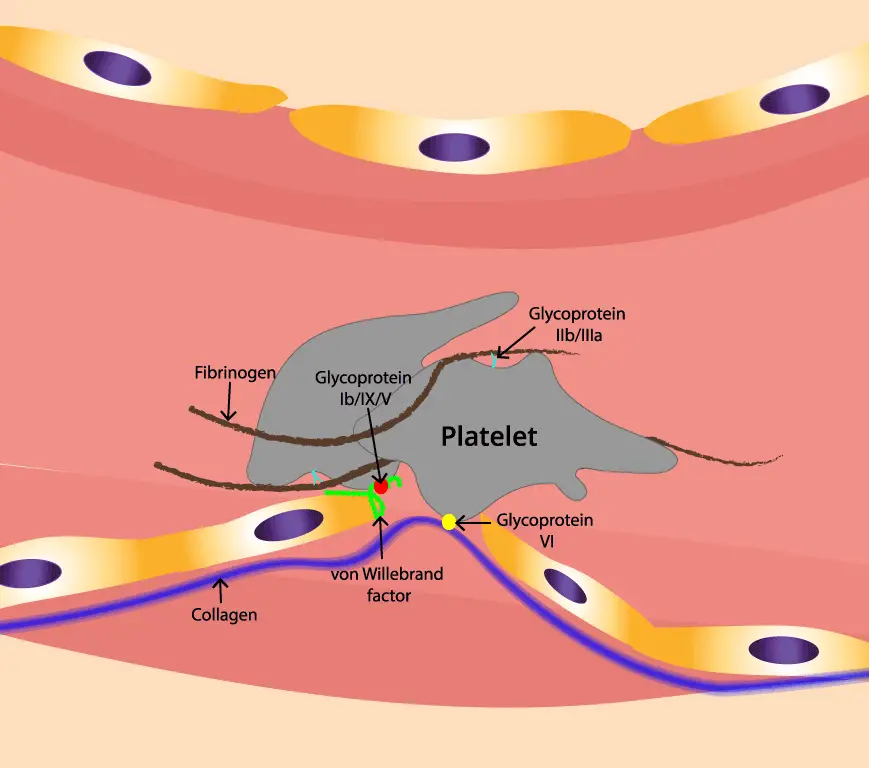
Why Multiple Adhesions Matter
Each glycoprotein has its own strengths and limitations:
- GPIb-IX: Fast and reversible, ideal for initial tethering but not strong enough for stable adhesion.
- Integrins: Powerful and long-lasting, but take time to activate and unfold.
By working together, these glycoproteins overcome their individual weaknesses and create a robust adhesion network. GPIb-IX’s initial grab slows down and positions the platelet, allowing integrins to take over and form the strong, permanent bonds needed for a stable clot.
Other Players
Beyond the key players, other adhesive molecules like selectins and CD40L contribute to platelet adhesion under specific conditions, adding further complexity and resilience.
Conformational and Cross-Linking Changes
Conformational and cross-linking changes are crucial for platelet adhesion and aggregation, the foundation of a stable clot.
Shape Shifting for a Grip
Platelets undergo dramatic shape changes during activation, driven by cytoskeletal rearrangements:
- Discoid to Spheroid: Upon receiving activation signals, the actin cytoskeleton reorganizes, causing the platelet to transform from a smooth disc to a spiky sphere. This increases the surface area for adhesion and inter-platelet interactions.
- Pseudopod Protrusion: Actin filaments form finger-like extensions called pseudopods, further maximizing the platelet’s grip on the exposed subendothelial matrix and neighboring platelets.
Integrins
Integrins, the key adhesion molecules, undergo conformational changes as platelets activate:
- Bent to Extended: Initially, integrins lie in a bent, inactive conformation. Upon activation, they undergo a “switchblade-like” transition, extending their legs and revealing binding sites for ligands like fibrinogen and collagen.
- Cluster Formation: Activated integrins cluster together, increasing their binding affinity and forming strong adhesion points.
Fibrinogen
Fibrinogen, a vital clotting factor, acts as a cross-linker between activated platelets:
- Fibrinogen Binding: The extended integrins on activated platelets bind to specific sites on fibrinogen molecules.
- Bridging the Gap: Fibrinogen, with its two “arms,” connects multiple platelets, forming a fibrinogen-platelet meshwork that stabilizes the clot.
The Interplay for Efficiency
These dynamic changes work in concert to facilitate efficient platelet adhesion and aggregation:
- Shape shift first: Early shape changes increase platelet contact with the matrix, bringing integrins closer to potential binding sites.
- Integrin activation follows: Activation signals trigger integrin conformational changes, exposing their binding sites.
- Fibrinogen joins the party: Activated integrins readily bind fibrinogen, bridging the gap between platelets and solidifying the clot structure.
Therapeutic Implications
Understanding these processes opens doors for targeted therapies:
- Antiplatelet drugs: Inhibiting integrin activation or preventing fibrinogen binding can hinder clot formation in thrombotic disorders.
- Promoting clot formation: Drugs mimicking fibrinogen’s bridge-building function can be used to address bleeding diatheses.
Von Willebrand Factor (vWF)
vWF, with its diverse abilities, orchestrates the critical stages of platelet adhesion and aggregation, ensuring hemostasis and preventing excessive bleeding.
vWF and GPIb-IX
The initial encounter between platelets and the injured vessel wall is mediated by a delicate interaction between vWF and GPIb-IX:
- vWF Anchors: vWF binds to collagen fibers exposed in the subendothelial matrix, acting as an adhesive anchor for platelets.
- GPIb-IX Recognition: The A1 domain of vWF interacts with the α chain of GPIb-IX on the platelet surface, slowing its flow and initiating the adhesion process.
- Catch and Release: This tethering is weak and transient, allowing the platelet to roll along the vWF-coated collagen fibers, searching for stronger adhesion points.
The Rolling Rhythm
This rolling motion isn’t just random; it’s a crucial step in platelet activation:
- Shear Flow Resistance: vWF’s elongated structure acts as a tether, resisting the high shear forces of blood flow and preventing the platelet from being washed away.
- Signaling Symphony: During rolling, GPIb-IX transmits signals into the platelet, initiating internal activation processes like shape change and granule release.
- Integrin Activation: This rolling process exposes the platelet to collagen and other activating stimuli, preparing it for stronger adhesion through integrins.
vWF and the Bridging Role
When the platelet finds a suitable collagen binding site:
- Integrin Engagement: Activated αIIbβ3 integrins on the platelet bind directly to collagen, providing a strong and stable adhesion point.
- vWF Bridge: Its multimeric structure allows it to bind both collagen and the platelet’s integrins, acting as a molecular bridge between them.
- Platelet Cluster Formation: This vWF-mediated bridging facilitates the binding of additional platelets, forming a growing and stable platelet aggregate, the foundation of the clot.
Therapeutic Implications
Understanding vWF’s diverse functions opens doors for targeted therapies:
- vWF-based therapies: Recombinant vWF or vWF-mimetic drugs can be used to promote adhesion and address bleeding diatheses.
- Antiplatelet drugs: Inhibiting vWF-GPIb-IX or vWF-integrin interactions can prevent unwanted clot formation in thrombotic disorders.
Platelet Aggregation Mechanisms
Platelet aggregation, the process by which platelets clump together, forms the foundation of blood clot formation (hemostasis). This intricate process plays a critical role in preventing excessive bleeding after injury, but when uncontrolled, can lead to dangerous thrombus formation. Understanding the mechanisms of platelet aggregation is crucial for managing both bleeding and thrombotic disorders.
The Activated Platelet
Platelet activation is triggered by stimuli like collagen exposure or ADP release. This activation triggers a cascade of events:
- Shape Change: The platelet transforms from a smooth disc to a spiky sphere, maximizing its surface area for adhesion.
- Granule Release: Potent hemostatic molecules like ADP and serotonin are released, amplifying the activation signal and attracting other platelets.
- Integrin Activation: The key player, GPIIb-IIIa integrin, undergoes conformational changes, revealing binding sites for fibrinogen.
Fibrinogen Bridges
Fibrinogen, a large plasma protein, acts as the crucial bridge between activated platelets:
- Fibrinogen Binding: Activated GPIIb-IIIa receptors on one platelet bind to specific sites on fibrinogen molecules.
- Bridging the Gap: The elongated structure of fibrinogen allows it to stretch and bind to integrins on another platelet, forming a molecular bridge.
- Growing Network: As more platelets join the dance, fibrinogen bridges link them together, creating a growing and stable platelet aggregate.
GPIIb-IIIa Activation and Signaling
GPIIb-IIIa activation plays a pivotal role in the aggregation process:
- Calcium Influx: Increased calcium levels within the platelet activate GPIIb-IIIa, promoting its conformational change and exposing fibrinogen binding sites.
- Positive Feedback Loop: Binding of fibrinogen to GPIIb-IIIa further activates the integrin, leading to its clustering and increased affinity for fibrinogen, amplifying the aggregation cascade.
- Signaling Pathways: GPIIb-IIIa activation triggers downstream signaling pathways that contribute to granule release, shape change, and further integrin activation, perpetuating the aggregation process.
Additional Players
While fibrinogen and GPIIb-IIIa take center stage, other molecules contribute to the intricate process:
- Thromboxane A2: This potent vasoconstrictor and platelet activator amplifies the aggregation process by stimulating integrin activation and granule release.
- ADP and Thrombin: These released molecules activate other platelets, propagating the aggregation signal and increasing the size and stability of the clot.
- vWF: While not directly involved in bridging, vWF plays a crucial role in initial tethering and rolling of platelets on the injured vessel wall, facilitating their interaction with fibrinogen and GPIIb-IIIa.
Thrombus Formation and Resolution
The culmination of this process is the formation of a stable thrombus, a platelet plug sealing the injured vessel wall and preventing blood loss:
- Dense Network: Fibrinogen bridges continually connect activated platelets, forming a dense and stable clot structure.
- Fibrin Reinforcement: As coagulation factors cascade, fibrin polymers further reinforce the clot, providing additional strength and stability.
- Clot Resolution: Once hemostasis is achieved, fibrinolytic pathways activate, gradually degrading the clot to restore normal blood flow.
Therapeutic Implications
Understanding the mechanisms of platelet aggregation holds immense therapeutic potential:
- Antiplatelet drugs: Targeting various steps in the process, like GPIIb-IIIa antagonists or ADP receptor inhibitors, can prevent unwanted clot formation in thrombotic disorders.
- Promoting clot formation: Mimicking fibrinogen or inhibiting platelet disaggregation can aid in addressing bleeding diatheses.
Regulation of Aggregation
Platelet aggregation, while vital for hemostasis, needs to be tightly regulated to prevent unwanted clot formation. There are various mechanisms that act as brakes and counterpoints to maintain the delicate balance of platelet activation and aggregation.
Nitric Oxide (NO)
- Guanylate cyclase activation: NO stimulates an enzyme called guanylate cyclase in platelets, leading to increased production of cyclic guanosine monophosphate (cGMP).
- cGMP-mediated inhibition: cGMP dampens platelet activation by inhibiting calcium mobilization, shape change, and granule release, ultimately reducing aggregation.
- Vascular relaxation: NO also relaxes blood vessels, promoting smooth blood flow and further hindering unwanted clot formation.
Prostacyclin
- cAMP elevation: Prostacyclin stimulates adenylate cyclase in platelets, leading to higher levels of cyclic adenosine monophosphate (cAMP).
- cAMP-mediated inhibition: Similar to cGMP, cAMP inhibits platelet activation and aggregation by suppressing calcium signaling and other downstream pathways.
- Endothelial production: Prostacyclin is primarily produced by endothelial cells lining the blood vessels, acting as a local antiplatelet shield.
Disintegrins
- GPIIb-IIIa blockade: Disintegrins prevent fibrinogen binding to the GPIIb-IIIa integrin, the key receptor for platelet aggregation.
- Fibrinogen dissociation: Some disintegrins can even disrupt existing fibrinogen bridges, breaking down already formed platelet aggregates.
The Interplay of the Players
These anti-aggregation mechanisms cooperate in a coordinated manner:
- NO and prostacyclin work synergistically: Both molecules elevate cGMP and cAMP levels, amplifying their inhibitory effects on platelet activation.
- Endothelial cells orchestrate the flow: Prostacyclin production by endothelial cells creates a localized antiplatelet environment near the vessel wall, preventing unwanted clot formation while allowing platelets to aggregate at injury sites.
- Disintegrins offer targeted intervention: In therapeutic settings, disintegrins can be used to specifically block GPIIb-IIIa and prevent clot formation in conditions like acute coronary syndromes.
Clinical Implications
Understanding these regulatory mechanisms opens doors for therapeutic approaches:
- Nitric oxide donors: Drugs that release NO can be used to improve blood flow and reduce platelet aggregation in various cardiovascular disorders.
- Prostacyclin analogs: Synthetic forms of prostacyclin can be administered to enhance antiplatelet activity and prevent thrombosis.
- Disintegrin-based therapies: Drugs like abciximab, a GPIIb-IIIa antagonist, find application in preventing clot formation during angioplasty procedures.
Thrombopoiesis
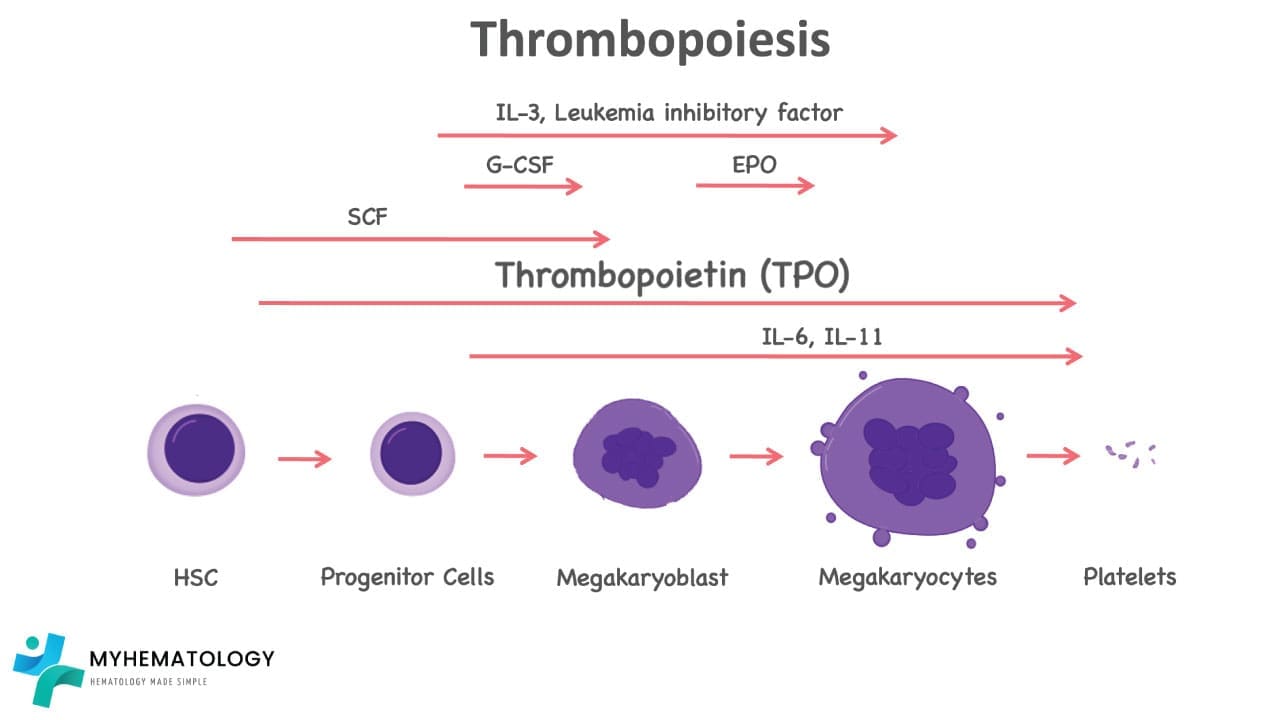
The process of platelet production
- Commitment: The journey begins with hematopoietic stem cells. Under the influence of specific growth factors, these pluripotent cells commit to the megakaryocytic lineage.
- Proliferation: Committed megakaryocyte progenitors undergo multiple rounds of cell division, multiplying their numbers and preparing for the grand transformation.
- Maturation: As they mature, megakaryocytes begin to grow in size, becoming the largest cells in the bone marrow. Their nuclei undergo endomitosis, replicating their DNA content without cell division, leading to multilobular nuclei with up to 128 lobes.
- Cytoplasmic expansion: The cytoplasm of the megakaryocyte balloons, filled with ribosomes and organelles dedicated to protein synthesis. An elaborate network of microtubules forms, providing structural support for the upcoming transformation.
- Platelet demarcation: Within the cytoplasm, unique structures called demarcation channels begin to form. These are complex invaginations of the plasma membrane, pinching off platelet-sized portions of the cytoplasm containing all the necessary machinery for platelet function.
- Platelet release: As demarcation channels mature, they sever completely, releasing fully formed platelets into the bone marrow sinusoids, ready to circulate in the bloodstream.
Cytokines and Growth Factors in Thrombopoiesis
Thrombopoiesis is tightly regulated by cytokines and growth factors.
Thrombopoietin (TPO)
Thrombopoietin is the key cytokine in thrombopoiesis. Produced by the liver and kidney, TPO binds to its specific receptor (c-Mpl) on megakaryocyte progenitors and mature megakaryocytes, triggering a cascade of signals:
- Proliferation: TPO stimulates the division and expansion of megakaryocyte progenitors, ensuring a healthy reserve of these cells ready for differentiation.
- Maturation: TPO orchestrates the complex process of megakaryocyte maturation, leading to the formation of the multi-lobed nuclei and cytoplasmic expansion crucial for platelet production.
- Platelet demarcation: TPO plays a critical role in the formation and maturation of platelet demarcation channels, ensuring the efficient and precise release of functional thrombocytes.
Other Cytokines and Growth Factors
- Interleukin-3 (IL-3): This versatile cytokine synergizes with TPO, enhancing megakaryocyte proliferation and differentiation, especially in emergency situations requiring rapid platelet production.
- Interleukin-6 (IL-6): Similar to IL-3, IL-6 supports TPO signaling by promoting megakaryocyte proliferation and survival.
- Stem cell factor (SCF): This growth factor works in concert with TPO to maintain the pool of megakaryocyte progenitors in the bone marrow.
- Insulin-like growth factor-1 (IGF-1): This ubiquitous growth factor contributes to megakaryocyte maturation and platelet production via its stimulatory effects on cell growth and survival.
Interplay and Regulation
These cytokines and growth factors don’t play in isolation; they interact and modulate each other’s effects:
- Synergy: TPO can amplify the effects of IL-3 and IL-6, resulting in a more robust proliferative response.
- Feedback Loops: Megakaryocyte-derived factors can feed back to the liver and kidney, influencing TPO production and maintaining a balanced system.
- Inhibition: Other molecules like interferon-alpha can act as brakes, suppressing thrombopoiesis when platelet levels are sufficient.
Therapeutic Implications
Understanding the symphony of cytokines and growth factors in thrombopoiesis opens doors for:
- Treating thrombocytopenia: Recombinant TPO or TPO mimetics can stimulate platelet production in conditions like chemotherapy-induced thrombocytopenia or aplastic anemia.
- Managing autoimmune thrombocytopenia: Understanding the role of cytokines in platelet destruction can guide the development of targeted therapies for this condition.
- Fine-tuning coagulation disorders: Modulating specific cytokine pathways might offer novel strategies for managing both bleeding and thrombotic disorders.
Causes of High Platelet Count (Thrombocytosis)
Thrombocytosis, a condition characterized by an abnormally high thrombocyte count, can be classified into two primary types: primary and secondary.
Primary Thrombocytosis
- Definition: This type arises from an abnormality within the bone marrow, specifically in the megakaryocytes, which produce platelets.
- Causes:
- Essential thrombocythemia (ET): A myeloproliferative disorder where the bone marrow overproduces thrombocytes leading to high platelet count.
- Familial thrombocytosis: A genetic condition that leads to excessive platelet production.
Secondary Thrombocytosis
- Definition: This type is a result of an underlying condition or stimulus that triggers the body to produce more thrombocoytes leading to high platelet count.
- Common Causes:
- Inflammation: Conditions like rheumatoid arthritis, systemic lupus erythematosus, and inflammatory bowel disease can stimulate platelet production.
- Infection: Viral or bacterial infections, especially those that cause inflammation, can lead to increased thrombocyte count.
- Malignancy: Cancers, particularly those affecting the bone marrow or blood cells, can cause secondary thrombocytosis.
- Iron deficiency: A deficiency of iron can sometimes lead to increased platelet production.
- Post-splenectomy: Removal of the spleen, which filters old platelets, can result in a temporary increase in thrombocyte count.
- Medications: Certain medications, such as corticosteroids and anti-inflammatory drugs, can stimulate platelet production.
Thrombocytosis is often associated with an increased risk of bleeding and it can also lead to an increased risk of thrombosis (blood clot formation). The specific risks and management strategies depend on the underlying cause and the severity of the condition. Thrombocytosis can be a sign of an underlying condition but it doesn’t always cause symptoms. In some cases, it may be an incidental finding on a blood test.
Causes of Low Platelets (Thrombocytopenia)
Thrombocytopenia is a condition characterized by an abnormally low platelet count. It can be classified into three main types: peripheral destruction, decreased production, and sequestration.
Peripheral Destruction
- Definition: This occurs when platelets are destroyed at a faster rate than they are produced leading to low platelets.
- Common Causes:
- Immune thrombocytopenia (ITP): An autoimmune disorder where the body’s immune system attacks and destroys platelets.
- Drug-induced thrombocytopenia: Certain medications can cause the immune system to attack platelets causing low platelets.
- Infections: Viral or bacterial infections can lead to platelet destruction.
- Disseminated intravascular coagulation (DIC): A life-threatening condition where blood clots form throughout the body, consuming platelets.
Decreased Production
- Definition: This occurs when the bone marrow fails to produce enough thrombocytes.
- Common Causes:
- Aplastic anemia: A rare condition where the bone marrow stops producing blood cells.
- Bone marrow infiltration: Conditions like leukemia, lymphoma, or metastatic cancer can infiltrate the bone marrow and interfere with platelet production.
- Inherited bone marrow disorders: Certain inherited conditions can affect platelet production.
- Nutritional deficiencies: Deficiencies in vitamins or minerals can impair bone marrow function.
Sequestration
- Definition: This occurs when thrombocytes are trapped in the spleen or other organs leading to low platelets available.
- Common Causes:
- Hypersplenism: An enlarged spleen that traps blood cells, including platelets.
- Portal hypertension: Increased pressure in the portal vein, which can lead to splenomegaly and platelet sequestration.
Pseudo-Thrombocytopenia
- Definition: Pseudo-thrombocytopenia is a laboratory artifact where an automated blood counter reports a falsely low platelet count in a patient who actually has a normal number of circulating thrombocytes. Misdiagnosing a patient with true thrombocytopenia can lead to unnecessary, expensive, and potentially invasive investigations or treatments (like steroid therapy or splenectomy).
- Common Causes:
- EDTA-Dependent Agglutination: EDTA-dependent agglutination is triggered when the anticoagulant chelates calcium, causing a conformational change in the platelet surface glycoprotein IIb/IIIa that exposes “cryptic” antigens to which pre-existing autoantibodies bind, resulting in the formation of platelet clumps.
- Platelet Satellitism: This is a rare phenomenon where platelets adhere to the surface of neutrophils (and sometimes other white blood cells) in an EDTA sample. The analyzer counts the neutrophil-platelet complex as a single white blood cell, again lowering the platelet count.
- Giant Platelets: Conditions like Bernard-Soulier syndrome or May-Hegglin anomaly produce platelets so large that the analyzer counts them as red blood cells or white blood cells.
- Cold Agglutinins: Sometimes, antibodies that react at room temperature or colder can cause clumping.
- Clotted Specimen: If the blood draw was difficult or the tube was not mixed properly immediately after collection, a small micro-clot may form. This consumes platelets, leading to a low count. This is technically a “pre-analytical error” rather than classic biological PCT, but the result is the same: a false low count.
Laboratory Investigations for Platelet Evaluation
Several laboratory tests are used to evaluate platelet function and count. These tests help in diagnosing and monitoring platelet disorders.
Complete Blood Count (CBC)
- Purpose: A CBC provides a comprehensive assessment of blood cells, including platelets.
- Parameters:
- Platelet count: The number of platelets per microliter of blood.
- Reference range: 150,000 – 450,000 platelets/µL
- Mean platelet volume (MPV): The average size of platelets.
- Reference range: 7.5 – 10.5 µm³
- Platelet distribution width (PDW): The variation in platelet size.
- Reference range: 10 – 15%
- Platelet count: The number of platelets per microliter of blood.
Platelet Function Tests
- Bleeding time: Measures the time it takes for a standardized skin incision to stop bleeding.
- Reference range: 2-9 minutes
- Platelet aggregation studies: Assess platelet aggregation in response to various agonists (e.g., collagen, adenosine diphosphate [ADP], epinephrine).
- Results: Evaluated based on the degree of platelet aggregation.
Bone Marrow Examination
- Purpose: To evaluate platelet production in the bone marrow and identify any underlying disorders.
- Procedure: A bone marrow sample is obtained through a needle biopsy and examined under a microscope.
- Findings: Can reveal abnormalities in megakaryocyte development or other bone marrow disorders.
Coagulation Tests
- Prothrombin time (PT): Measures the clotting time of plasma in the extrinsic pathway.
- Reference range: 11-13.5 seconds
- International normalized ratio (INR): Standardizes PT results across different laboratories.
- Reference range: 0.8-1.2
- Activated partial thromboplastin time (aPTT): Measures the clotting time of plasma in the intrinsic pathway.
- Reference range: 25-35 seconds
Note: The reference ranges for these tests may vary slightly depending on the laboratory and the specific patient population. It’s important to interpret these results in conjunction with other clinical findings and the patient’s medical history.
Clinical Implications of Platelet Disorders
Platelet disorders can have significant clinical implications, particularly in relation to bleeding and thrombotic complications.
Bleeding Complications
- Mucosal bleeding: Nosebleeds, bleeding gums, and easy bruising are common manifestations of thrombocytopenia.
- Cutaneous bleeding: Petechiae (small, reddish-purple spots) and ecchymoses (bruises) can occur due to decreased platelet count.
- Gastrointestinal bleeding: Ulceration and bleeding in the gastrointestinal tract can be severe complications of thrombocytopenia.
- Menorrhagia: Excessive menstrual bleeding can be a symptom of platelet disorders.
Thrombotic Complications
- Thrombosis: Platelet disorders, especially those with elevated platelet counts (thrombocytosis), can increase the risk of blood clot formation (thrombosis) in blood vessels.
- Deep vein thrombosis (DVT): Blood clots in the deep veins of the legs can lead to pain, swelling, and potentially pulmonary embolism.
- Pulmonary embolism: A blood clot that travels to the lungs can cause chest pain, shortness of breath, and even death.
- Cerebral thrombosis: Blood clots in the brain can lead to stroke and neurological damage.
- Coronary artery disease: Increased platelet aggregation can contribute to the formation of atherosclerotic plaques in the coronary arteries, increasing the risk of heart attack.
The specific clinical implications of platelet disorders can vary depending on the underlying cause and the severity of the platelet abnormality.
Management of Platelet Disorders
The management of platelet disorders depends on the underlying cause and the severity of the condition. Treatment may involve medications, supportive care, or a combination of both.
Medical Management
- Corticosteroids: Often used to treat immune-mediated thrombocytopenia, such as ITP. They can help suppress the immune system and increase platelet production.
- Immunosuppressants: Medications like cyclophosphamide, azathioprine, or vincristine may be used in more severe cases of ITP or other autoimmune disorders.
- Rituximab: A monoclonal antibody that targets B cells, may be effective in treating ITP, especially in patients who are resistant to corticosteroids.
- Thrombopoietin receptor agonists: These drugs stimulate the production of platelets by activating the thrombopoietin receptor. They can be used to treat thrombocytopenia due to bone marrow disorders or certain chemotherapy regimens.
- Platelet transfusions: In cases of severe thrombocytopenia with active bleeding, platelet transfusions may be necessary to temporarily increase the platelet count.
Supportive Care
- Avoidance of unnecessary procedures: Invasive procedures that can increase the risk of bleeding should be minimized in patients with thrombocytopenia.
- Hemostatic agents: Topical agents like desmopressin or tranexamic acid may be used to help control bleeding in some cases.
- Monitoring: Regular monitoring of platelet count and other blood tests is essential to assess the effectiveness of treatment and detect any complications.
- Lifestyle modifications: Patients with platelet disorders may need to avoid activities that increase the risk of injury or bleeding, such as contact sports or strenuous exercise.
The management of platelet disorders requires a multidisciplinary approach involving hematologists, primary care physicians, and other specialists. The specific treatment plan will depend on the individual patient’s condition, symptoms, and overall health.
Frequently Asked Questions
What is a dangerously low platelet count?
While normal ranges are 150,000–450,000/µL, spontaneous bleeding typically does not occur until counts drop below 20,000/µL. Surgical bleeding concerns may arise below 50,000/µL. However, the “dangerous” level depends on the individual patient and the cause of the low count.
Can stress cause high platelets?
Yes. This is known as “reactive” or secondary thrombocytosis. Physical or physiological stress (like surgery, trauma, or severe infection) can cause a temporary release of stored platelets from the spleen and stimulate new production.
What is the lifespan of a platelet?
Platelets circulate in the blood for approximately 7 to 10 days. After this period, they are removed from circulation by the spleen and liver.
Why do patients with liver disease often have low platelets?
Liver disease causes thrombocytopenia through two main mechanisms:
1) The liver produces less Thrombopoietin (TPO), reducing platelet production, and
2) Portal hypertension causes the spleen to enlarge (splenomegaly), which traps (sequesters) platelets, removing them from circulation.
What is the difference between aspirin and other blood thinners regarding platelets?
Aspirin is an antiplatelet agent that specifically stops platelets from clumping by inhibiting Thromboxane A2. Other “blood thinners” like warfarin or heparin are anticoagulants that work on clotting factors (proteins) in the plasma, not directly on the platelets themselves.
Glossary of Related Medical Terms
- Adhesion: The initial process where platelets stick to the injured vessel wall (specifically collagen and von Willebrand factor).
- Aggregation: The process where platelets stick to each other to form a plug, primarily mediated by fibrinogen bridges.
- Alpha Granules: The most abundant platelet granules containing proteins like fibrinogen, von Willebrand factor, and Factor V.
- Dense Granules: Smaller granules containing small molecules like ADP, ATP, Serotonin, and Calcium.
- Glycocalyx: The outer sugar-rich coating of the platelet that prevents inappropriate adhesion to healthy endothelium.
- Hemostasis: The physiological process of stopping bleeding.
- Megakaryocyte: The large bone marrow cell responsible for producing thrombocytes.
- Petechiae: Pinpoint, round spots that appear on the skin as a result of bleeding; a common sign of thrombocytopenia.
- Pseudopods: “False feet” or extensions of the cytoplasm that platelets project when activated to grab onto surfaces.
- Thrombocytopenia: A medical condition characterized by an abnormally low platelet count (<150,000/µL).
- Thrombocytosis: A medical condition characterized by an abnormally high platelet count (>450,000/µL).
- Thrombopoietin (TPO): The primary hormone regulating thrombocyte production, produced mainly by the liver.
Disclaimer: This article is intended for informational purposes only and is specifically targeted towards medical students. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- de Gaetano G. Historical overview of the role of platelets in hemostasis and thrombosis. Haematologica. 2001 Apr;86(4):349-56. PMID: 11325638.
- Gibbins JM, Mahaut-Smith MP. Platelets and Megakaryocytes: Volume 1: Functional Assays (Humana Press). 2004.
- Estevez B, Du X. New Concepts and Mechanisms of Platelet Activation Signaling. Physiology (Bethesda). 2017 Mar;32(2):162-177. doi: 10.1152/physiol.00020.2016. PMID: 28228483; PMCID: PMC5337829.
- Li Z, Delaney MK, O’Brien KA, Du X. Signaling during platelet adhesion and activation. Arterioscler Thromb Vasc Biol. 2010 Dec;30(12):2341-9. doi: 10.1161/ATVBAHA.110.207522. Epub 2010 Nov 11. PMID: 21071698; PMCID: PMC3085271.
- Andrews RK, López JA, Berndt MC. Molecular mechanisms of platelet adhesion and activation. Int J Biochem Cell Biol. 1997 Jan;29(1):91-105. doi: 10.1016/s1357-2725(96)00122-7. PMID: 9076944.
- Rumbaut RE, Thiagarajan P. Platelet-Vessel Wall Interactions in Hemostasis and Thrombosis. San Rafael (CA): Morgan & Claypool Life Sciences; 2010.
- Ruggeri ZM and Mendolicchio GL. Adhesion Mechanisms in Platelet Function. Circulation Research. 2007;100:1673–1685.
- Kaushansky K. Historical review: megakaryopoiesis and thrombopoiesis. Blood. 2008 Feb 1;111(3):981-6. doi: 10.1182/blood-2007-05-088500. PMID: 18223171; PMCID: PMC2214745.
- Schulze H, Shivdasani RA. Mechanisms of thrombopoiesis. Journal of Thrombosis and Haemostasis. 2005; 3(8): 1717-1724.
- Noh JY. Megakaryopoiesis and Platelet Biology: Roles of Transcription Factors and Emerging Clinical Implications. Int J Mol Sci. 2021 Sep 5;22(17):9615. doi: 10.3390/ijms22179615. PMID: 34502524; PMCID: PMC8431765.