Key Takeaways
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare, acquired clonal hematopoietic stem cell disorder characterized by complement-mediated hemolysis.
- PIGA gene mutation leading to deficiency of GPI-anchored proteins (CD55, CD59).
- Uncontrolled complement activation, causing intravascular hemolysis and thrombosis.
- Hemolytic anemia (fatigue, dark urine, jaundice). Hemoglobinuria is hallmark for PNH.
- Thrombosis (abdominal pain, hepatic/cerebral vein thrombosis).
- Bone marrow failure (pancytopenia, infections).
- Other symptoms: abdominal pain, esophageal spasm, erectile dysfunction.
- Flow cytometry (gold standard, detects CD55/CD59 deficiency).
- CBC (anemia, reticulocytosis, pancytopenia).
- LDH elevation (hemolysis).
- Hemoglobinuria (dark urine).
- Negative DAT.
- Supportive care (transfusions, folic acid).
- Complement inhibitors (eculizumab, ravulizumab).
- Bone marrow transplantation (curative, for severe cases).
- Anticoagulation.
- Monitoring for complications.
- Thrombosis (major cause of mortality).
- Renal failure.
- Pulmonary hypertension.
- Evolution to MDS/AML.
- Improved significantly with complement inhibitors.
- Dependent on disease severity and complications.
*Click ▾ for more information
Introduction
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare, acquired blood disorder characterized by the destruction of red blood cells by the body's complement system. Paroxysmal nocturnal hemoglobinuria (PNH) is not an inherited condition but rather an acquired disease that develops during a person's lifetime. The origin of this disorder lies within the bone marrow, specifically in the hematopoietic stem cells (HSCs), which are the precursor cells responsible for the production of all mature blood cells.
For medical students, understanding paroxysmal nocturnal hemoglobinuria (PNH) is crucial due to its potential for severe complications such as thrombosis and kidney failure if left undiagnosed and untreated . While paroxysmal nocturnal hemoglobinuria (PNH) is rare, its diverse and often non-specific initial symptoms necessitate a high level of clinical suspicion in certain patient presentations.
Why does paroxysmal nocturnal hemoglobinuria cause nocturnal hemoglobinuria?
A point of historical interest, and potential confusion for students, lies in the name "nocturnal" in paroxysmal nocturnal hemoglobinuria (PNH). This term originated from the observation that some patients noticed their urine was darker in the morning . However, it is now understood that the hemolysis associated with paroxysmal nocturnal hemoglobinuria (PNH) occurs continuously throughout the day and night. The increased concentration of urine during sleep can make the presence of hemoglobin, released from the destroyed red blood cells, more apparent in the first void of the morning.
What age is paroxysmal nocturnal hemoglobinuria (PNH) diagnosed?
Paroxysmal nocturnal hemoglobinuria (PNH) can manifest at any age, but it is most frequently diagnosed in young adults, typically between the ages of 30 and 40 years. Interestingly, some studies suggest that women are slightly more likely to develop PNH than men.
Pathophysiology of Paroxysmal Nocturnal Hemoglobinuria (PNH)
PIGA Gene Mutation
Paroxysmal nocturnal hemoglobinuria (PNH) originates from an acquired somatic mutation in the phosphatidylinositol glycan class A (PIGA) gene. This mutation arises spontaneously within a single hematopoietic stem cell residing in the bone marrow.
The PIGA gene is located on the X chromosome and is crucial for the synthesis of glycosylphosphatidylinositol (GPI). This X-linked location influences how the mutation expresses itself.
In males, who possess only one X chromosome (XY), a mutation in their single copy of the PIGA gene in a hematopoietic stem cell will lead to the development of paroxysmal nocturnal hemoglobinuria (PNH) in all blood cells derived from that mutated cell.
In females, who have two X chromosomes (XX), one of the X chromosomes in each somatic cell is randomly inactivated early in embryonic development, a process known as lyonization. If the active X chromosome in a hematopoietic stem cell carries the PIGA mutation, the resulting clone of blood cells will also exhibit the paroxysmal nocturnal hemoglobinuria (PNH) defect. The mutated stem cell then undergoes proliferation, giving rise to a population of abnormal blood cells, all carrying the same PIGA gene defect. This process establishes paroxysmal nocturnal hemoglobinuria (PNH) as a clonal disorder.
Deficiency of GPI-Anchored Proteins
The GPI anchor is a complex glycolipid structure that acts as a tether, attaching various proteins to the surface membrane of blood cells. In paroxysmal nocturnal hemoglobinuria (PNH) individuals, the mutation in the PIGA gene results in a non-functional or significantly impaired enzyme. This leads to a deficiency or complete absence of the GPI anchor on the surface of the affected blood cells .
Several crucial proteins that normally protect blood cells from the body's own immune system are anchored to the cell surface via this GPI anchor. In paroxysmal nocturnal hemoglobinuria (PNH), these protective proteins are either missing or significantly reduced on the affected cells.
The most clinically significant of these are CD55 (decay-accelerating factor) and CD59 (membrane inhibitor of reactive lysis or protectin). These two proteins play a vital role in regulating the activity of the complement system.
Uncontrolled Complement Activation
The complement system is an integral part of the innate immune system, acting as a crucial defense mechanism against invading pathogens. It comprises a cascade of plasma proteins that, when activated, can directly destroy microorganisms, enhance phagocytosis, and promote inflammation.
Under normal physiological conditions, CD55 and CD59 serve as critical regulators, preventing the complement system from attacking and destroying the body's own healthy blood cells. In paroxysmal nocturnal hemoglobinuria (PNH), the deficiency of these GPI-anchored regulatory proteins on the surface of red blood cells leaves these cells vulnerable to uncontrolled activation of the complement system.
This leads to chronic, inappropriate complement-mediated destruction of red blood cells within the bloodstream, resulting in intravascular hemolysis. The subsequent breakdown of red blood cells releases hemoglobin into the circulation, which is responsible for many of the clinical manifestations and complications observed in paroxysmal nocturnal hemoglobinuria (PNH). The complement activation also activates platelets, and other procoagulant factors, leading to an increased risk of blood clot formation.
The consequences of these pathophysiological processes include:
- Anemia: Due to the destruction of RBCs.
- Hemoglobinuria: Hemoglobin in the urine, often most noticeable in the morning.
- Thrombosis: Blood clots in various parts of the body, particularly in unusual locations like the hepatic and mesenteric veins.
- Organ damage: Due to chronic hemolysis and thrombosis.
Signs and Symptoms of Paroxysmal Nocturnal Hemoglobinuria (PNH)
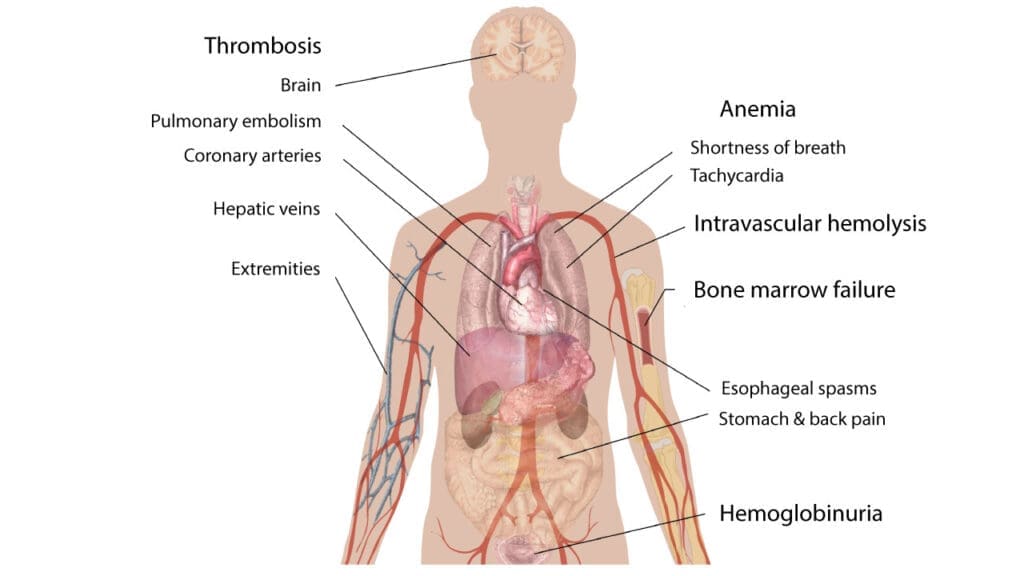
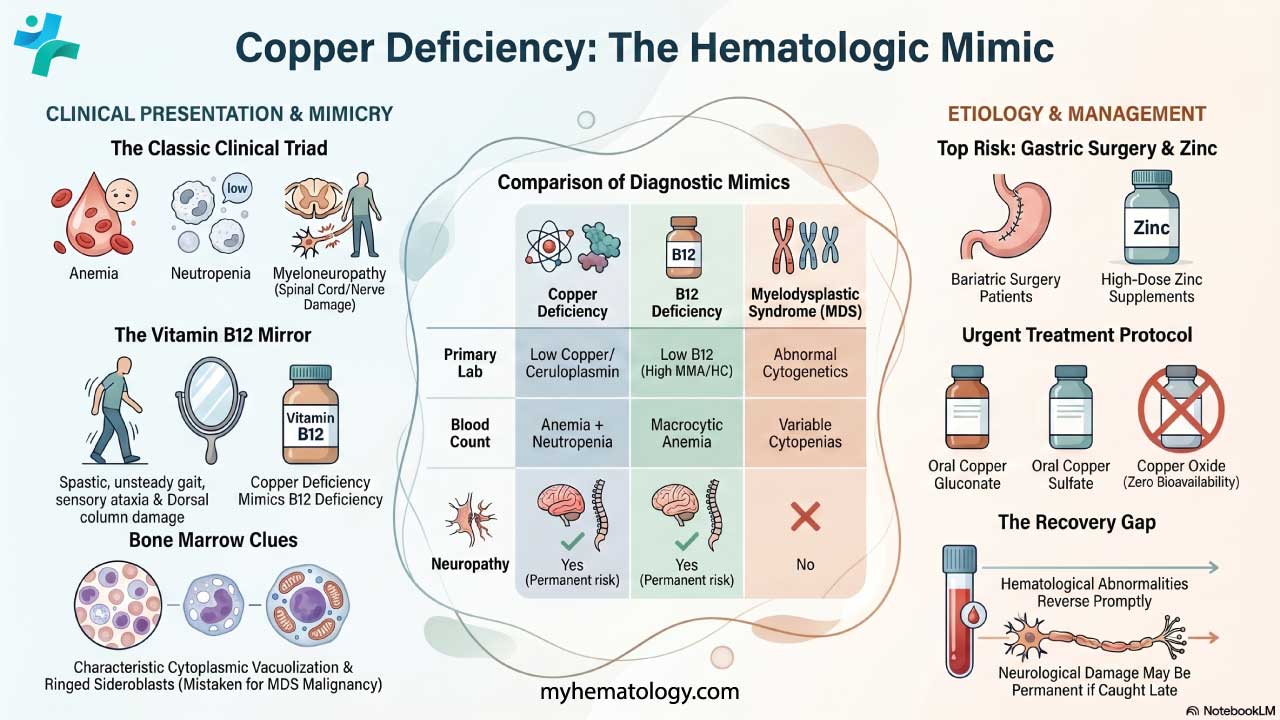
Paroxysmal nocturnal hemoglobinuria (PNH) presents with a wide range of signs and symptoms, which can vary significantly from person to person. This variability makes diagnosis challenging.
Classic Triad
When discussing Paroxysmal Nocturnal Hemoglobinuria (PNH), the "classic triad" refers to a set of three key clinical manifestations that have historically been associated with the disease. It's important to note that while these three features are significant, not every paroxysmal nocturnal hemoglobinuria (PNH) patient will present with all of them.
- Hemolytic Anemia: This is a core feature of paroxysmal nocturnal hemoglobinuria (PNH). It involves the destruction of red blood cells (hemolysis), leading to a decrease in their number. This results in symptoms like fatigue, weakness, pallor, and shortness of breath. A very characteristic sign of this hemolytic anemia is hemoglobinuria, which is the presence of hemoglobin in the urine, often causing it to appear dark or reddish-brown.
- Thrombosis: Paroxysmal nocturnal hemoglobinuria (PNH) creates a prothrombotic state, meaning it increases the risk of blood clots. These clots can occur in atypical locations, such as the hepatic veins (Budd-Chiari syndrome), mesenteric veins, and cerebral veins, which can lead to serious complications. Thrombosis is a major cause of morbidity and mortality in paroxysmal nocturnal hemoglobinuria (PNH) patients.
- Bone Marrow Failure: Paroxysmal nocturnal hemoglobinuria (PNH) can be associated with bone marrow dysfunction, which can manifest as pancytopenia (a deficiency of all three blood cell types: red blood cells, white blood cells, and platelets). This can lead to anemia, increased susceptibility to infections, and bleeding problems. It is important to understand that the relationship between bone marrow failure and paroxysmal nocturnal hemoglobinuria (PNH) is complex, and they can occur together.
Other Symptoms
A significant aspect of paroxysmal nocturnal hemoglobinuria (PNH) pathophysiology involves the depletion of nitric oxide (NO). Intravascular hemolysis releases free hemoglobin, which scavenges and inactivates NO, a vital molecule for smooth muscle relaxation and vascular tone. This NO depletion can lead to several characteristic symptoms:
- Dysphagia and esophageal spasms (difficulty or pain swallowing due to smooth muscle constriction in the esophagus)
- Abdominal pain (caused by spasms of the smooth muscles in the gastrointestinal tract),
- Back pain
- Erectile dysfunction in male patients (due to impaired vasodilation in the penis) .
Laboratory Investigations in Confirming the Diagnosis
The diagnosis of PNH relies on a combination of clinical suspicion and specific laboratory investigations.
Complete Blood Count
Initial screening tests often include a complete blood count (CBC), which may reveal anemia (low hemoglobin levels) and possible pancytopenia (low counts of all blood cell types) if bone marrow failure is present.
The reticulocyte count, a measure of new red blood cell production, is usually elevated as the bone marrow attempts to compensate for the ongoing hemolysis, although it might be lower than expected in cases with associated bone marrow failure.
Biochemical tests
Serum markers of intravascular hemolysis are also crucial. Serum lactate dehydrogenase (LDH) is typically markedly elevated, while serum haptoglobin, a protein that binds free hemoglobin, is usually low or absent . The indirect bilirubin level may also be mildly elevated due to red blood cell breakdown .
Urinalysis
Urinalysis can provide further clues, potentially revealing hemoglobinuria (hemoglobin in the urine), which can cause the characteristic dark urine. In chronic cases, hemosiderinuria (iron deposits in the urine) might also be detected.
Flow Cytometry
This is the most sensitive and specific test for paroxysmal nocturnal hemoglobinuria (PNH). It detects the absence or reduced expression of GPI-anchored proteins, particularly CD55 and CD59, on blood cell surfaces.
The fluorescently labeled aerolysin (FLAER) assay is particularly valuable as it binds directly to the GPI anchor and is highly sensitive for detecting GPI deficiency on granulocytes and monocytes.
Flow cytometry can also quantify the paroxysmal nocturnal hemoglobinuria (PNH) clone size, which is the percentage of blood cells lacking these proteins. It is important to analyze multiple blood cell lineages, including red blood cells and granulocytes/monocytes, as red blood cell transfusions can mask the paroxysmal nocturnal hemoglobinuria (PNH) clone in the red blood cell population .
Other laboratory tests
- Peripheral Blood Smear: Possible presence of schistocytes (fragmented red blood cells), indicating hemolysis.
- Direct Antiglobulin Test (DAT or Coombs Test): Negative DAT, as paroxysmal nocturnal hemoglobinuria (PNH) is not an autoimmune disorder.
- Bone Marrow Examination: May show increased cellularity. It can help identify concurrent bone marrow failure syndromes. It is used when contemplating bone marrow transplant.
Older tests like the acidified serum lysis test (Ham test) and the sucrose lysis test (sugar water test), which demonstrated increased complement sensitivity of paroxysmal nocturnal hemoglobinuria (PNH) red blood cells, are now less commonly used due to the superior sensitivity and specificity of flow cytometry .
| Test | Typical Findings | Significance |
| Complete Blood Count (CBC) | Anemia (low hemoglobin), potentially low WBCs and platelets | Initial assessment of blood cell counts |
| Reticulocyte Count | Elevated (compensatory), but may be lower than expected for anemia severity | Assesses bone marrow response to hemolysis |
| Serum Lactate Dehydrogenase (LDH) | Markedly elevated | Indicator of intravascular hemolysis |
| Serum Haptoglobin | Low or absent | Binds free hemoglobin; depleted in hemolysis |
| Serum Bilirubin (Indirect) | May be elevated | Product of red blood cell breakdown |
| Urinalysis | Hemoglobinuria, hemosiderinuria | Detects hemoglobin and iron deposits in urine due to hemolysis |
| Flow Cytometry for GPI-anchored proteins (CD55, CD59, FLAER) | Absence or deficiency of GPI-anchored proteins on RBCs, granulocytes, monocytes | Gold standard for diagnosis, quantifies PNH clone size |
Treatment and Management of Paroxysmal Nocturnal Hemoglobinuria (PNH)
The treatment and management of Paroxysmal Nocturnal Hemoglobinuria (PNH) have evolved significantly, particularly with the advent of complement inhibitors. The management of Paroxysmal Nocturnal Hemoglobinuria (PNH) involves a combination of supportive care, targeted therapies aimed at the complement system, and in rare cases, hematopoietic stem cell transplantation.
Supportive care
Supportive care includes blood transfusions to manage anemia and alleviate symptoms. Iron and folic acid supplementation may be necessary to support red blood cell production. Anticoagulants are used to prevent and treat thrombotic events .
Complement Inhibitors
The advent of complement inhibitors has significantly improved the prognosis for paroxysmal nocturnal hemoglobinuria (PNH) patients.
- Eculizumab (Soliris®) is a monoclonal antibody that inhibits the C5 protein in the terminal complement pathway, preventing red blood cell lysis.
- Ravulizumab-cwvz (Ultomiris®) is another C5 inhibitor with a longer half-life, requiring less frequent administration.
- Newer complement inhibitors include pegcetacoplan (Empaveli®), a C3 inhibitor ; iptacopan (Fabhalta™), an oral factor B inhibitor; crovalimab (Piasky®), another C5 inhibitor with subcutaneous administration; and danicopan, a factor D inhibitor used in specific cases.
Bone Marrow Transplantation
Hematopoietic stem cell transplantation (HSCT) is the only potentially curative therapy, especially for younger patients but is reserved for severe cases due to its risks.
Others
- Anticoagulation: Prophylaxis and treatment of thrombosis includes warfarin, low molecular weight heparin, and direct oral anticoagulants (DOACs).
- Immunosuppressive therapy: For paroxysmal nocturnal hemoglobinuria (PNH) with concurrent aplastic anemia.
Long-Term Management and Monitoring of PNH
Long-term management involves regular monitoring of blood counts, LDH levels, and paroxysmal nocturnal hemoglobinuria (PNH) clone size via flow cytometry. Strategies to prevent and manage complications such as thrombosis, renal disease, pulmonary hypertension, iron deficiency, and infections are crucial.
Patient education regarding their condition, treatment adherence, and awareness of potential complications is essential. The management often requires a multidisciplinary approach and the prognosis for patients with paroxysmal nocturnal hemoglobinuria (PNH) has significantly improved with the advent of complement inhibitors .
Complications and Prognosis
Paroxysmal Nocturnal Hemoglobinuria (PNH), if left untreated or poorly managed, can lead to a range of severe complications that significantly impact prognosis.
Complications
- Thrombosis: This is the most significant cause of morbidity and mortality in paroxysmal nocturnal hemoglobinuria (PNH). Blood clots can occur in unusual locations, such as the hepatic veins (Budd-Chiari syndrome), mesenteric veins, and cerebral veins. Thrombotic events can lead to organ damage, including liver failure, bowel infarction, and stroke.
- Renal Failure: Chronic hemoglobinuria can damage the kidneys, leading to renal insufficiency and, in severe cases, renal failure. Iron deposition in the kidneys can also contribute to renal damage.
- Pulmonary Hypertension: Chronic hemolysis can lead to pulmonary hypertension, a condition characterized by high blood pressure in the pulmonary arteries. This can cause shortness of breath, chest pain, and heart failure.
- Myelodysplastic Syndrome (MDS) and Acute Myeloid Leukemia (AML): Paroxysmal nocturnal hemoglobinuria (PNH) can evolve into MDS or AML, both of which are serious blood cancers. This risk is higher in patients with concurrent bone marrow failure.
- Iron Overload: Chronic transfusions can lead to iron overload, which can damage various organs, including the liver, heart, and endocrine glands.
- Infections: Patients receiving compliment inhibitors are at an increased risk of meningococcal infection. Patients with bone marrow failure have increased risk of infections due to leukopenia.
Prognosis
The prognosis of paroxysmal nocturnal hemoglobinuria (PNH) has improved significantly with the advent of complement inhibitors like eculizumab and ravulizumab. These drugs have dramatically reduced the incidence of thrombosis and improved overall survival. However, paroxysmal nocturnal hemoglobinuria (PNH) remains a chronic and potentially life-threatening disease.
- Factors Affecting Prognosis
- Severity of hemolysis.
- Presence and severity of thrombosis.
- Development of complications, such as renal failure or pulmonary hypertension.
- Evolution to MDS or AML.
- Response to treatment.
- Access to appropriate treatment.
- Impact of New Treatments: Complement inhibitors have significantly extended the life expectancy of paroxysmal nocturnal hemoglobinuria (PNH) patients.
However, long-term monitoring is essential to manage potential complications and ensure optimal outcomes. Bone marrow transplant, while curative, carries significant risks, and is not appropriate for all patients. It is important to understand that early diagnosis and appropriate management are crucial for improving the prognosis of paroxysmal nocturnal hemoglobinuria (PNH).
Frequently Asked Questions (FAQs)
What is the hallmark of paroxysmal nocturnal hemoglobinuria (PNH)?
The hallmark of PNH is hemoglobinuria, particularly dark or reddish-brown urine, often most noticeable in the morning. While other symptoms like anemia and thrombosis are significant, the presence of hemoglobin in the urine due to the breakdown of red blood cells is a very characteristic and often defining feature of the disease.
Who is at risk for paroxysmal nocturnal hemoglobinuria (PNH)?
While PNH can affect individuals of any age, race, or gender, there are some factors that appear to be associated with an increased risk or are found in conjunction with PNH.
- History of aplastic anemia
- Other bone marrow disorders like MDS and AML
- Although PNH can occur at any age, it is most often diagnosed in people in their 30s and 40s.
What is the life expectancy with PNH?
Though historically associated with reduced life expectancy, modern complement inhibitors have significantly improved patient outcomes, allowing for near-normal life expectancy with appropriate management.
Can paroxysmal nocturnal hemoglobinuria (PNH) turn into leukemia?
Yes, there is a recognized, albeit relatively low, risk of PNH evolving into acute myeloid leukemia (AML). While PNH itself is not a leukemia, it is a bone marrow disorder, and the instability within the bone marrow can, in some cases, lead to the development of AML. This risk is a factor that clinicians consider when managing PNH patients, and it underscores the importance of ongoing monitoring.
Disclaimer: This article is intended for informational purposes only and is specifically targeted towards medical students. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Bektas M, Copley-Merriman C, Khan S, Sarda SP, Shammo JM. Paroxysmal nocturnal hemoglobinuria: role of the complement system, pathogenesis, and pathophysiology. J Manag Care Spec Pharm. 2020 Dec;26(12-b Suppl):S3-S8. doi: 10.18553/jmcp.2020.26.12-b.s3. PMID: 33356782; PMCID: PMC10408413.
- Devalet B, Mullier F, Chatelain B, Dogné JM, Chatelain C. Pathophysiology, diagnosis, and treatment of paroxysmal nocturnal hemoglobinuria: a review. Eur J Haematol. 2015 Sep;95(3):190-8. doi: 10.1111/ejh.12543. Epub 2015 Mar 26. PMID: 25753400.
- Bektas M, Copley-Merriman C, Khan S, Sarda SP, Shammo JM. Paroxysmal nocturnal hemoglobinuria: current treatments and unmet needs. J Manag Care Spec Pharm. 2020 Dec;26(12-b Suppl):S14-S20. doi: 10.18553/jmcp.2020.26.12-b.s14. PMID: 33356783; PMCID: PMC10410676.
- Brodsky RA. How I treat paroxysmal nocturnal hemoglobinuria. Blood. 2021 Mar 11;137(10):1304-1309. doi: 10.1182/blood.2019003812. PMID: 33512400; PMCID: PMC7955407.
- Bektas M, Copley-Merriman C, Khan S, Sarda SP, Shammo JM. Paroxysmal nocturnal hemoglobinuria: patient journey and burden of disease. J Manag Care Spec Pharm. 2020 Dec;26(12-b Suppl):S8-S14. doi: 10.18553/jmcp.2020.26.12-b.s8. PMID: 33356781; PMCID: PMC10408416.