Key Takeaways
Systemic Mastocytosis is a clonal myeloproliferative neoplasm characterized by the abnormal accumulation of neoplastic mast cells in extracutaneous organs, most commonly the bone marrow.
- Molecular Driver ▾: The KIT D816V somatic mutation is the hallmark of the disease, found in over 90% of adult cases, leading to constitutive activation of the KIT receptor.
- WHO Subtypes ▾: The disease ranges from Indolent Systemic Mastocytosis (normal life expectancy) to Advanced variants like Aggressive Systemic Mastocytosis and Mast Cell Leukemia, which require cytoreductive or targeted therapy.
- Clinical Presentation ▾: Patients present with a "dual" clinical picture: mediator-release symptoms (anaphylaxis, flushing, GI distress) and organ infiltration symptoms (cytopenias, hepatosplenomegaly, and bone damage).
- Diagnostic Gold Standard ▾: A definitive diagnosis requires a bone marrow biopsy demonstrating multifocal mast cell aggregates (Major Criterion) or a combination of minor criteria including atypical morphology, CD25/CD30 expression, KIT mutation, and elevated serum tryptase.
- Modern Management ▾: Treatment has shifted toward precision medicine using targeted KIT inhibitors like Avapritinib and Midostaurin, alongside traditional symptom management (antihistamines and trigger avoidance).
*Click ▾ for more information
Introduction
Systemic Mastocytosis is a rare, clonal myeloproliferative neoplasm (MPN) characterized by the abnormal proliferation, survival, and accumulation of mast cells within extracutaneous organs. Unlike reactive mast cell activation, which is a transient response to an allergen, Systemic Mastocytosis represents a permanent, neoplastic change where mast cells accumulate uncontrollably, often due to a specific genetic "on switch" (the KIT mutation).
While mast cells are most famously known for their role in Type I hypersensitivity (allergic) reactions, in Systemic Mastocytosis, they infiltrate tissues such as the bone marrow, liver, spleen, and gastrointestinal tract.
Classification Context
It is crucial to distinguish Systemic Mastocytosis from Cutaneous Mastocytosis. While Cutaneous Mastocytosis is largely limited to the skin and is more common in children, Systemic Mastocytosis involves internal organs and is predominantly diagnosed in adults. Because of its rarity, affecting roughly 1 in 10,000 people, it is classified as an orphan disease, frequently requiring a high index of clinical suspicion to diagnose.
Etiology and Pathophysiology
The Molecular Driver: The KIT Mutation
The primary cause of Systemic Mastocytosis is a somatic gain-of-function mutation in the KIT gene (located on chromosome 4q12). This gene encodes a Type III receptor tyrosine kinase (KIT, also known as CD117) expressed on the surface of mast cells and their precursors.
In over 90% of adult patients, a specific point mutation occurs where the amino acid aspartate (D) is replaced by valine (V) at position 816 in the phosphotransferase domain (Exon 17).
Normally, the KIT receptor requires the binding of its ligand, Stem Cell Factor (SCF), to dimerize and activate. The D816V mutation causes the receptor to be "locked" in an active state. This leads to ligand-independent, continuous signaling.
This is almost always a somatic mutation (acquired in hematopoietic stem cells) rather than a germline mutation (inherited).
Downstream Signaling and Cellular Effects
The hyperactive KIT receptor triggers several intracellular signaling pathways that drive the neoplastic process:
- PI3K / AKT / mTOR: Promotes mast cell survival and inhibits apoptosis (programmed cell death).
- JAK / STAT: Enhances proliferation and differentiation of mast cell progenitors.
- MAPK / ERK: Drives further proliferation and cellular growth.
The result is an accumulation of "immortal" mast cells that do not die off at the normal rate and continue to multiply, eventually forming the characteristic dense aggregates seen in bone marrow biopsies.
The Mechanism of Mediator Release
Pathophysiology in Systemic Mastocytosis isn't just about cell numbers; it’s about cell activity. Neoplastic mast cells have a lower threshold for degranulation.
- Primary Mediators (Pre-stored): Histamine, heparin, and various proteases (specifically tryptase and chymase).
- Secondary Mediators (Newly synthesized): Prostaglandin D2 (PGD2) and leukotrienes (LTC4).
These mediators act on various receptors (H1, H2) throughout the body, causing the diverse systemic symptoms ranging from gastric acid hypersecretion to life-threatening anaphylaxis.
Tissue Infiltration and Organ Dysfunction
In the more aggressive forms of the disease (ASM, MCL), the pathophysiology shifts from "mediator-driven" to "infiltration-driven."
- Bone Marrow: Mast cell aggregates replace normal hematopoietic tissue, leading to cytopenias (anemia, thrombocytopenia).
- Skeleton: Mast cells produce factors that influence bone remodeling (like RANKL and IL-6), leading to localized osteolytic lesions or generalized osteoporosis and osteosclerosis.
- Gastrointestinal / Hepatic: Infiltration of the liver and spleen causes hepatosplenomegaly, portal hypertension, and malabsorption.
WHO Classification of Systemic Mastocytosis
The current classification (WHO 5th Edition/ICC) relies heavily on the presence of "B-findings" (indicators of high disease burden) and "C-findings" (indicators of organ dysfunction/cytoreduction requirement).
B-Findings (Burden of disease)
- High Mast Cell Burden: Bone marrow biopsy showing > 30% infiltration by mast cells and/or serum tryptase > 200 ng/mL.
- Signs of Dysplasia: Evidence of hypercellularity or myelodysplasia in non-mast cell lineages (but not enough to diagnose a separate neoplasm).
- Organomegaly: Hepatomegaly or splenomegaly without impaired organ function.
C-Findings (Cytoreduction-requiring / Organ Damage)
- Cytopenias: Absolute Neutrophil Count (ANC) < 1 x 109/L, Hemoglobin <10 g/dL, or Platelets < 100 x 109/L.
- Hepatopathy: Palpable hepatomegaly with impaired liver function, ascites, or portal hypertension.
- Skeletal Involvement: Pathological fractures or large osteolytic lesions.
- Gastrointestinal: Malabsorption with weight loss due to GI infiltration.
- Splenomegaly: With associated hypersplenism.
The Five Main WHO Subtypes
The classification divides Systemic Mastocytosis into "Non-advanced" and "Advanced" variants.
I. Indolent Systemic Mastocytosis (ISM)
- Criteria: Meets general criteria for Systemic Mastocytosis; has fewer than two B-findings and zero C-findings.
- Clinical Picture: The most common form. Patients typically suffer from mediator-release symptoms (skin flushing, GI distress) but have a near-normal life expectancy.
- Bone Marrow: Low-level infiltration.
II. Smoldering Systemic Mastocytosis (SSM)
- Criteria: Meets general criteria for Systemic Mastocytosis; has two or more B-findings but zero C-findings.
- Clinical Picture: A "bridge" between indolent and aggressive disease. There is a higher risk of progression to advanced Systemic Mastocytosis.
III. Systemic Mastocytosis with an Associated Hematologic Neoplasm (SM-AHN)
- Criteria: The patient meets the criteria for Systemic Mastocytosis and meets the WHO criteria for another distinct blood disorder (e.g., Chronic Myelomonocytic Leukemia (CMML), Myelodysplastic Syndrome (MDS), or an Acute Myeloid Leukemia (AML)).
- Clinical Picture: Prognosis is usually determined by the "associated" neoplasm, which is often aggressive.
IV. Aggressive Systemic Mastocytosis (ASM)
- Criteria: Meets general criteria for Systemic Mastocytosis and has at least one C-finding.
- Clinical Picture: This is a high-risk variant. The mast cells are physically damaging organs. Patients require cytoreductive therapy or targeted KIT inhibitors (like Avapritinib or Midostaurin).
V. Mast Cell Leukemia (MCL)
- Criteria: A rare and highly aggressive variant. Mast cells must make up ≥ 20% of cells on a bone marrow aspirate smear.
- Leukemic Phase: If mast cells represent ≥ 10% of peripheral blood white cells, it is classified as "classic" MCL; if not, it is "aleukemic" MCL.

Summary Table
| Subtype | B-Findings | C-Findings | AHN Present? | Prognosis |
| Indolent (ISM) | 0 or 1 | None | No | Excellent |
| Smoldering (SSM) | ≥ 2 | None | No | Guarded/Variable |
| SM-AHN | Variable | Variable | Yes | Poor (usually based on AHN) |
| Aggressive (ASM) | Variable | ≥ 1 | No | Poor |
| Leukemia (MCL) | Variable | Variable | No | Very Poor |
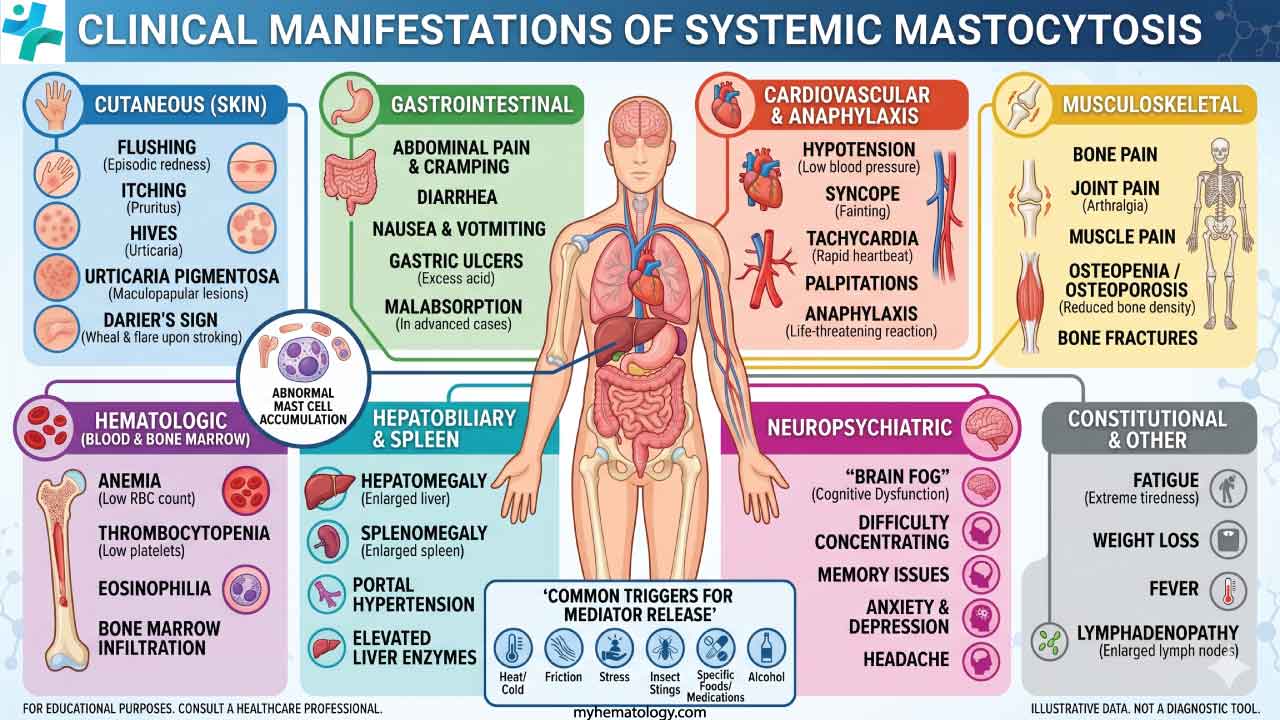
Clinical Manifestations of Systemic Mastocytosis
Clinical manifestations of Systemic Mastocytosis are remarkably diverse because mast cells are distributed throughout the body's connective tissues. We can categorize these symptoms into two distinct mechanisms: mediator-related symptoms (the "chemical storm") and organ infiltration symptoms (the "physical burden").
Mediator-Release Symptoms (Systemic Effects)
These symptoms occur when mast cells degranulate, releasing histamine, tryptase, prostaglandins, and leukotrienes into the circulation. These can be chronic or occur in paroxysmal "attacks" triggered by heat, exercise, stress, alcohol, or certain medications.
- Constitutional: Fatigue, weight loss, and fever.
- Skin: Episodic flushing (often the most common symptom), pruritus (itching), and urticaria.
- Gastrointestinal: Abdominal pain, cramping, nausea, vomiting, and chronic diarrhea. Gastric acid hypersecretion (due to histamine) can lead to peptic ulcer disease.
- Cardiovascular/Neurological: Hypotension, tachycardia, syncope (fainting), and "brain fog" or headaches.
- Anaphylaxis: Patients with Systemic Mastocytosis have a significantly higher risk of severe, unexplained anaphylaxis, particularly in response to Hymenoptera (wasp/bee) stings.
Organ Infiltration Symptoms (Advanced SM)
In aggressive variants, the symptoms are caused by the physical crowding out of normal tissue by mast cell aggregates (the "C-findings").
- Hematologic: Anemia (fatigue), thrombocytopenia (easy bruising/bleeding), or leukopenia (increased infection risk) due to bone marrow replacement.
- Hepatosplenic: Palpable hepatomegaly and splenomegaly. In advanced cases, this leads to portal hypertension, ascites, and impaired liver function.
- Gastrointestinal Infiltration: Malabsorption and protein-losing enteropathy, leading to significant weight loss and nutritional deficiencies.
Cutaneous Involvement
While the disease is systemic, the skin is involved in about 80% of Indolent Systemic Mastocytosis cases (though it is less frequent in advanced forms).
- Maculopapular Cutaneous Mastocytosis (MCM): Historically called Urticaria Pigmentosa, these are reddish-brown, hyperpigmented macules or papules.
- Darier’s Sign: A pathognomonic clinical sign. When a skin lesion is stroked or rubbed, it urticates (becomes itchy, red, and swollen) due to localized mast cell degranulation.
Skeletal and Bone Symptoms
Mast cells in the bone marrow microenvironment release cytokines (like RANKL and IL-6) that disrupt the balance between osteoblasts and osteoclasts.
- Bone Pain: Often described as deep, aching pain.
- Pathological Fractures: Due to severe osteoporosis or localized osteolytic lesions (bone "holes").
- Osteosclerosis: Paradoxically, some patients may show areas of increased bone density.
Summary of Clinical Correlation
| Organ System | Manifestation | Primary Mechanism |
| Skin | Flushing, Darier's sign, Pruritus | Mediator Release |
| GI Tract | Diarrhea, Peptic Ulcers, Malabsorption | Mediator + Infiltration |
| Bone Marrow | Cytopenias (Anemia, etc.) | Infiltration |
| Skeletal | Osteoporosis, Bone Pain | Cytokine-driven remodeling |
| Vascular | Hypotension, Anaphylaxis, Syncope | Histamine/Prostaglandins |
Laboratory Investigations & Diagnostics
The investigation of Systemic Mastocytosis is designed to fulfill the WHO diagnostic criteria while simultaneously assessing the "B" (burden) and "C" (complication) findings.
Serum Biomarkers: The Screening Phase
- Basal Serum Tryptase (BST): This is the most important initial screening test. A persistent basal level >20 ng/mL is a minor diagnostic criterion. In patients with advanced Systemic Mastocytosis, tryptase levels can exceed 200 ng/mL. However, some patients with Indolent Systemic Mastocytosis may have "normal" tryptase levels, so a low result does not 100% exclude the disease.
- Hereditary Alpha-Tryptasemia (HαT) Adjustment: Recent guidelines emphasize checking for HαT (a common genetic trait) if tryptase is mildly elevated, as this can cause a "false positive" minor criterion. The tryptase level should be adjusted using the formula: Adjusted BST = BST/(1 + extra TPSAB1 copies).
The Gold Standard: Bone Marrow Evaluation
A bone marrow aspirate and trephine biopsy are mandatory for a definitive diagnosis.
- Morphology (The Major Criterion): We look for multifocal, dense infiltrates of mast cells (aggregates of ≥ 15 mast cells). Neoplastic mast cells often appear atypical—spindle-shaped or elongated—rather than the normal round shape.
- Immunohistochemistry (IHC): To visualize the aggregates, we stain for:
- Tryptase: Marks all mast cells.
- CD117 (KIT): Marks the receptor; often shows high intensity in SM.
- CD25: The most reliable marker for "aberrant" (neoplastic) mast cells.
Flow Cytometry
Flow cytometry is used on the bone marrow aspirate to identify the "aberrant immunophenotype" of the mast cells. Neoplastic mast cells express markers that normal mast cells do not:
- CD25: Highly sensitive and the "classic" marker.
- CD2: Often present but less sensitive than CD25.
- CD30: A newer, highly useful marker that helps distinguish Systemic Mastocytosis from other myeloid neoplasms.
Molecular and Genetic Testing
- KIT D816V Mutation Analysis: This is a minor diagnostic criterion.
- High-Sensitivity Assays: Because the "clone" (percentage of mutated cells) can be very small, we use highly sensitive techniques like Digital PCR (dPCR) or Next-Generation Sequencing (NGS) rather than standard Sanger sequencing.
- Specimen: Can be performed on bone marrow or peripheral blood.
- Extended Myeloid Panels: In cases of suspected SM-AHN (Advanced Systemic Mastocytosis), we test for additional mutations in genes like SRSF2, ASXL1, and RUNX1. These are known as "S/A/R" mutations and signify a much poorer prognosis.
Assessment of Organ Involvement (B & C Findings)
Once the diagnosis is confirmed, we must "stage" the patient's organ function:
- Hematology: CBC to check for cytopenias (anemia, thrombocytopenia).
- Bone Health: DEXA Scan is essential for all Systemic Mastocytosis patients, as many develop premature osteoporosis or osteopenia due to mast cell-derived cytokines.
- Imaging:
- Abdominal Ultrasound or CT: To look for hepatosplenomegaly or lymphadenopathy.
- Skeletal X-rays/MRI: If the patient has bone pain, to look for osteolytic lesions or diffuse osteosclerosis.
- Gastrointestinal: Endoscopy and biopsy may be required if the patient has severe malabsorption or weight loss, looking for mast cell infiltration of the GI mucosa.
The Diagnostic Algorithm
- Elevated Tryptase? High suspicion.
- Peripheral Blood KIT Mutation? If positive, strongly suggests Systemic Mastocytosis.
- Bone Marrow Biopsy? Confirm with Morphology (aggregates) + IHC/Flow (CD25+).
- Organ Check? DEXA, CBC, and Liver function to determine subtype.
WHO Diagnostic Criteria for Systemic Mastocytosis (SM)
To establish the diagnosis of Systemic Mastocytosis, either one (1) major + one (1) minor criterion OR three (3) minor criteria must be fulfilled.
| Category | Criterion | Description |
| Major | Infiltration | Multifocal, dense infiltrates of mast cells (≥ 15 mast cells in an aggregate) detected in sections of bone marrow and/or other extracutaneous organ(s). |
| Minor 1 | Morphology | Atypical morphology: > 25% of the mast cells in the infiltrate are spindle-shaped or have an otherwise atypical morphology (e.g., hypogranulated), or > 25% of mast cells in bone marrow aspirate smears are immature/atypical. |
| Minor 2 | Molecular | KIT Mutation: Detection of an activating KIT point mutation at codon 816 (most commonly D816V) in bone marrow, blood, or another extracutaneous organ. |
| Minor 3 | Immunophenotype | Aberrant Markers: Mast cells in bone marrow, blood, or other extracutaneous organs express CD25, and/or CD2, and/or CD30 (standard mast cells are negative for these). |
| Minor 4 | Biomarker | Serum Tryptase: Persistent elevation of serum total tryptase > 20 ng/mL. Note: This criterion is not valid if the patient has an associated myeloid neoplasm (AHN). |
- The "3 Minor" Path: It is possible to diagnose Systemic Mastocytosis without seeing the large aggregates (the Major criterion) if the patient has the KIT mutation, the CD25 marker, and high tryptase. This is common in very early or "low-burden" Indolent Systemic Mastocytosis.
- The Tryptase Caveat: An elevated tryptase can be seen in other conditions (like Chronic Myeloid Leukemia or Hereditary Alpha-Tryptasemia). Therefore, in the presence of an associated myeloid neoplasm, the tryptase level loses its "minor criterion" status because it may be coming from the other cancer cells, not just the mast cells.
- The Importance of CD30: While CD25 is the most sensitive marker, the inclusion of CD30 in recent WHO updates is a significant diagnostic aid, especially when CD25 expression is weak.
Differential Diagnosis
The differential diagnosis of Systemic Mastocytosis (SM) is notoriously broad because the disease can mimic everything from simple food allergies to aggressive leukemias. When evaluating a patient for suspected Systemic Mastocytosis, clinicians must systematically rule out other conditions that cause mediator-related symptoms or unexplained organomegaly.
Mimics of Mediator-Related Symptoms (Flushing & GI Distress)
If a patient presents with episodic flushing, diarrhea, and hypotension, the following must be considered:
- Carcinoid Syndrome: Characterized by flushing and diarrhea caused by serotonin-secreting neuroendocrine tumors. Carcinoid flushing usually spares the limbs and is associated with right-sided heart valve disease. Diagnosis is confirmed by elevated 24-hour urinary 5-HIAA, whereas Systemic Mastocytosis will show elevated tryptase.
- Pheochromocytoma: Causes paroxysmal hypertension, palpitations, and sweating. Elevated urinary or plasma metanephrines. In Systemic Mastocytosis, hypotension (syncope) is more common than hypertension during an attack.
- Idiopathic Anaphylaxis: Recurrent episodes of anaphylaxis where no trigger is found. These patients lack the KIT D816V mutation and have normal bone marrow morphology.
- Hereditary Alpha-Tryptasemia (HαT): A common genetic trait where individuals have extra copies of the TPSAB1 gene. Patients have elevated baseline tryptase but do not have mast cell aggregates in the bone marrow or the KIT D816V mutation.
Mimics of Hematologic/Organ Infiltration
If the patient has hepatosplenomegaly or cytopenias, Systemic Mastocytosis must be distinguished from other myeloid neoplasms:
- Hypereosinophilic Syndrome (HES): Patients present with high eosinophil counts and organ damage. Look for the FIP1L1-PDGFRA fusion gene. While Systemic Mastocytosis can have associated eosinophilia, the presence of mast cell aggregates and CD25+ staining is specific to Systemic Mastocytosis.
- Chronic Myelomonocytic Leukemia (CMML): Characterized by persistent monocytosis. Bone marrow biopsy and flow cytometry will show monocyte expansion rather than neoplastic mast cell aggregates.
- Other Myeloproliferative Neoplasms (MPN): Such as Essential Thrombocythemia or Polycythemia Vera. Testing for JAK2, CALR, or MPL mutations.
Differential for Bone Manifestations
- Primary Osteoporosis: Especially in younger men or pre-menopausal women. A diagnosis of "idiopathic" osteoporosis in a young person should always trigger a serum tryptase test to rule out occult Indolent SM.
Comparison Table: SM vs. Common Mimics
| Feature | Systemic Mastocytosis (SM) | Carcinoid Syndrome | HαT |
| Primary Biomarker | Serum Tryptase (Elevated) | Urinary 5-HIAA (Elevated) | Serum Tryptase (Elevated) |
| Flushing Pattern | Often with pruritus/hives | Often with telangiectasia | Variable, often mild |
| Bone Marrow | Spindle-shaped aggregates | Normal (unless metastatic) | Normal mast cells |
| Molecular Marker | KIT D816V | None | TPSAB1 gene copies |
| Anaphylaxis Risk | High | Low | Moderate/High |
Treatment and Management
The treatment of Systemic Mastocytosis (SM) has undergone a paradigm shift in recent years, moving from purely supportive care to precision medicine with the advent of highly selective KIT inhibitors. Management is tailored based on the patient's WHO subtype, focusing on symptom control for indolent disease and cytoreduction for aggressive variants.
General Principles and Trigger Avoidance
The first line of defense for every Systemic Mastocytosis patient is the identification and avoidance of mast cell degranulation triggers.
- Common Triggers: Heat, sudden temperature changes, friction (Darier’s sign), emotional stress, alcohol, and physical exertion.
- Medication Precautions: Caution with NSAIDs, opioids (morphine/codeine), and certain anesthetic agents (e.g., neuromuscular blocking agents).
- Emergency Preparedness: All Systemic Mastocytosis patients, regardless of subtype, should carry two epinephrine auto-injectors due to the high risk of severe anaphylaxis.
Symptomatic (Anti-Mediator) Therapy
For Indolent (ISM) and Smoldering (SSM) patients, the goal is to neutralize the "chemical storm" released by mast cells.
- H1 and H2 Antihistamines: The cornerstone of therapy. High doses (up to 4x standard) of H1 blockers (e.g., Cetirizine) are often combined with H2 blockers (e.g., Famotidine) to control skin and gastric symptoms.
- Mast Cell Stabilizers: Cromolyn sodium is highly effective for gastrointestinal symptoms (cramping, diarrhea) but must be taken multiple times daily.
- Leukotriene Antagonists: Montelukast can help with respiratory symptoms and skin flushing.
- Omalizumab (Anti-IgE): Often used for patients with recurrent, unexplained anaphylaxis or those with high sensitivity to insect venom.
Targeted Therapy: The KIT Inhibitors
These drugs directly target the KIT D816V mutation.
- Avapritinib (Ayvakit): A highly selective KIT D816V inhibitor.
- In Advanced SM: Approved as a first-line treatment, showing superior overall survival and organ response compared to older therapies.
- In Indolent SM: Recently approved (at lower doses, e.g., 25mg) for patients with severe symptoms that do not respond to standard antihistamines.
- Midostaurin: A multi-kinase inhibitor active against both wild-type and mutant KIT. Used primarily in Advanced Systemic Mastocytosis variants (ASM, MCL).
Subtype-Specific Management Strategies
| Subtype | Primary Treatment Goal | Key Therapeutic Agents |
| Indolent (ISM) | Symptom Control & QoL | Antihistamines, Cromolyn, Low-dose Avapritinib |
| Smoldering (SSM) | Monitoring & Symptom Control | Similar to ISM; consider cytoreduction if B-findings progress |
| Advanced (ASM/MCL) | Cytoreduction & Survival | Avapritinib (First-line), Midostaurin, Cladribine |
| SM-AHN | Treat both neoplasms | Targeted therapy for SM + specific therapy for the AHN (e.g., AML induction) |
Supportive and Specialized Care
- Bone Health: Since mast cells trigger bone resorption, patients require regular DEXA scans. Treatment includes Calcium, Vitamin D, and Bisphosphonates (e.g., Zoledronic acid) or Denosumab for osteoporosis.
- Venom Immunotherapy (VIT): Mandatory for any Systemic Mastocytosis patient with a confirmed IgE-mediated allergy to bee or wasp stings, as these are often fatal without VIT.
- Cytoreductive Therapy: In cases where targeted inhibitors are unavailable or fail, Cladribine (2-CdA) or Interferon-alpha may be used to reduce the mast cell burden.
The Treatment Pyramid
- Base: Trigger avoidance + Epinephrine.
- Middle: Anti-mediator drugs (H1/H2, Cromolyn).
- Top (Advanced): Targeted KIT Inhibitors (Avapritinib/Midostaurin) ± Stem Cell Transplant for fit patients with MCL.
Prognosis and Follow-Up
Prognosis by Subtype
Prognosis in Systemic Mastocytosis is primarily determined by the WHO subtype.
- Indolent SM (ISM): Patients generally have a near-normal life expectancy. The primary challenge is not mortality, but rather the morbidity associated with chronic, debilitating symptoms and poor quality of life.
- Smoldering SM (SSM): Prognosis is variable. While patients may remain stable for years, they have a higher risk of progression to Advanced Systemic Mastocytosis than those with the indolent form.
- Advanced SM (ASM, SM-AHN, MCL): These subtypes carry a significantly poorer prognosis due to organ failure.
- ASM: Median survival has historically been around 3 - 5 years, though this is improving with newer targeted therapies.
- SM-AHN: Prognosis often depends on the severity of the associated blood cancer.
- MCL: The most aggressive form, with a median survival traditionally measured in months (approx. 2 - 6 months) if untreated.
Prognostic Scoring Systems
In clinical practice, we use validated scoring systems to risk-stratify patients beyond just their WHO subtype.
- MARS (Mutation-Adjusted Risk Score): Used for Advanced Systemic Mastocytosis. It considers age (> 60 years), hemoglobin (< 10 g/dL), platelets (< 100 x 109/L), and the presence of high-risk mutations (SRSF2, ASXL1, RUNX1).
- MAPS (Mayo Alliance Prognostic System): Evaluates both indolent and advanced variants, incorporating the WHO subtype, age, alkaline phosphatase levels, and adverse mutations.
- IPSM (International Prognostic Scoring System for Mastocytosis): Categorizes patients into low, intermediate, and high-risk groups based on clinical and laboratory parameters.
Longitudinal Follow-Up and Monitoring
Because Systemic Mastocytosis is a chronic and potentially progressive disease, lifelong monitoring is required.
- Symptom Assessment: Use of standardized tools like the Mastocytosis Control Test (MCT) or Quality of Life (QoL) questionnaires to track the impact of mediator symptoms.
- Laboratory Monitoring:
- Serum Tryptase: Checked every 6 - 12 months in stable patients; more frequently in advanced cases or during treatment. A rising tryptase can be an early sign of disease progression.
- Complete Blood Count (CBC) & Liver Function: To monitor for emerging "C-findings" (anemia, thrombocytopenia, or liver dysfunction).
- Bone Health: A DEXA scan should be repeated every 1 - 2 years depending on the baseline density and treatment.
- Molecular Monitoring: In patients on targeted therapy (e.g., Avapritinib), the KIT D816V Variant Allele Frequency (VAF) is monitored via high-sensitivity PCR to assess the depth of molecular response.
The Goal of Follow-Up
- Optimization: Ensure symptoms are controlled and the patient can function normally.
- Vigilance: Identify early signs of progression from Indolent to Advanced SM, such as a sudden rise in tryptase, new organomegaly, or dropping blood counts.
Frequently Asked Questions (FAQs)
Can a patient have Systemic Mastocytosis if their serum tryptase is normal?
Yes. While elevated tryptase (> 20 ng/mL) is a minor diagnostic criterion, some patients with low-burden Indolent Systemic Mastocytosis may have levels within the normal range. Diagnosis in these cases relies on other criteria like bone marrow morphology and the KIT mutation.
Is Systemic Mastocytosis a form of leukemia?
Not always. Systemic Mastocytosis is a broad category. While most cases are indolent and not considered leukemic, one specific subtype—Mast Cell Leukemia (MCL)—is an extremely aggressive form of leukemia.
Why is a DEXA scan mandatory for these patients?
Mast cells release cytokines (like RANKL and IL-6) that accelerate bone resorption. Patients with Systemic Mastocytosis are at a significantly higher risk for early-onset osteoporosis and pathological fractures, even if they have no other symptoms.
Can children get Systemic Mastocytosis?
Most children have Cutaneous Mastocytosis, which often regresses after puberty. Systemic Mastocytosis is predominantly an adult-onset disease.
How does the KIT D816V mutation change treatment?
The presence of this specific mutation makes the disease resistant to older tyrosine kinase inhibitors like Imatinib. However, it is the primary target for newer, more selective inhibitors like Avapritinib.
Are all triggers for mast cell degranulation the same for every patient?
No. Triggers are highly individual. Common ones include heat, alcohol, and certain medications, but patients are encouraged to keep a diary to identify their specific environmental or dietary "sparks."
Glossary of Related Medical Terms
- Anaphylaxis: A severe, potentially life-threatening systemic allergic reaction resulting from massive mast cell degranulation.
- B-Findings: Clinical or laboratory markers indicating a high burden of mast cells (e.g., tryptase >200 ng/mL) without evidence of actual organ dysfunction.
- C-Findings: Markers indicating "Cytoreduction-requiring" organ damage, such as cytopenias, portal hypertension, or pathological fractures.
- CD117: Also known as the KIT receptor; a surface marker used to identify mast cells via immunohistochemistry or flow cytometry.
- Clonal Disorder: A condition where a population of cells is derived from a single mutated progenitor cell.
- Darier Sign: A clinical finding where stroking a mast cell skin lesion causes it to become itchy and swollen (urticate).
- Degranulation: The process by which mast cells release pre-formed chemical mediators (like histamine) from their internal granules into the surrounding environment.
- Mast Cell: A type of white blood cell derived from myeloid stem cells that plays a key role in the innate immune response and allergic reactions.
- Myeloproliferative Neoplasm (MPN): A group of blood cancers where the bone marrow produces too many of one or more types of blood cells.
- Serum Tryptase: An enzyme found in mast cell granules; its baseline level in the blood serves as a primary biomarker for mast cell burden.
- Tyrosine Kinase Inhibitor (TKI): A type of targeted drug that blocks the action of enzymes responsible for cell signaling, growth, and division.
References
- Gangireddy M, Ciofoaia GA. Systemic Mastocytosis. [Updated 2023 Jul 4]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2026 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK544345/
- Valent, P., Akin, C., Sperr, W. R., Horny, H. P., Arock, M., Metcalfe, D. D., & Galli, S. J. (2023). New Insights into the Pathogenesis of Mastocytosis: Emerging Concepts in Diagnosis and Therapy. Annual review of pathology, 18, 361–386. https://doi.org/10.1146/annurev-pathmechdis-031521-042618
- Cilloni, D., Maffeo, B., Savi, A., Danzero, A. C., Bonuomo, V., & Fava, C. (2024). Detection of KIT Mutations in Systemic Mastocytosis: How, When, and Why. International journal of molecular sciences, 25(20), 10885. https://doi.org/10.3390/ijms252010885
- Wang, S. A., Orazi, A., Gotlib, J., Reiter, A., Tzankov, A., Hasserjian, R. P., Arber, D. A., & Tefferi, A. (2023). The international consensus classification of eosinophilic disorders and systemic mastocytosis. American journal of hematology, 98(8), 1286–1306. https://doi.org/10.1002/ajh.26966
- Madigan, L. M., Boggs, N. A., Rets, A. V., Gru, A. A., Tashi, T., Wada, D. A., Florell, S. R., & Carter, M. C. (2025). Mastocytosis in the Skin: Approach to Diagnosis, Evaluation, and Management in Adult and Pediatric Patients. American journal of clinical dermatology, 26(4), 499–510. https://doi.org/10.1007/s40257-025-00947-7
- Syal, A., Toh, J., McInerney, A., & Tremblay, D. (2025). The evaluation, management, and future of indolent systemic mastocytosis. Annals of hematology, 104(8), 3917–3927. https://doi.org/10.1007/s00277-025-06475-y
- Pardanani A. (2023). Systemic mastocytosis in adults: 2023 update on diagnosis, risk stratification and management. American journal of hematology, 98(7), 1097–1116. https://doi.org/10.1002/ajh.26962
- Nicolosi, M., Patriarca, A., Andorno, A., Mahmoud, A. M., Gennari, A., Boldorini, R., Gaidano, G., & Crisà, E. (2021). Precision Medicine in Systemic Mastocytosis. Medicina (Kaunas, Lithuania), 57(11), 1135. https://doi.org/10.3390/medicina57111135
- Gotlib, J., Reiter, A., & DeAngelo, D. J. (2022). Avapritinib for advanced systemic mastocytosis. Blood, 140(15), 1667–1673. https://doi.org/10.1182/blood.2021014612
- Ustun, C., Keklik Karadag, F., Linden, M. A., Valent, P., & Akin, C. (2025). Systemic mastocytosis: current status and challenges in 2024. Blood advances, 9(9), 2048–2062. https://doi.org/10.1182/bloodadvances.2024012612