Key Takeaways
Acute Lymphoblastic Leukemia (ALL) is a rapidly progressing malignancy of immature lymphoid cells (lymphoblasts) in the bone marrow. Acute lymphoblastic leukemia (ALL) is primarily classified by WHO as B-ALL (∼80% of cases) or T-ALL (∼15−20%).
- Pathophysiology ▾: The disease results from genetic abnormalities (translocations/mutations) that block B- or T-cell maturation and drive uncontrolled proliferation. Uncontrolled lymphoblast growth in the marrow causes bone marrow failure by suppressing normal blood cell production and subsequently extramedullary spread.
- Causes and Risk Factors ▾: A strong risk factor is having an inherited genetic syndrome, such as Down syndrome or ataxia-telangiectasia. Exposure to high doses of ionizing radiation is a known, though rare, risk factor.
- Symptoms ▾: Symptoms of bone marrow failure including anemia, thrombocytopenia and neutropenia. Common signs of infiltration include bone pain (marrow expansion), palpable hepatosplenomegaly, and lymphadenopathy.
- Laboratory Investigations and Diagnosis ▾: Diagnosis is confirmed by the presence of ≥20% lymphoblasts in the bone marrow aspirate or peripheral blood. While FISH and PCR identify key prognostic translocations (e.g., t(9;22),t(12;21)) for risk stratification. A Lumbar Puncture (LP) is mandatory at diagnosis to check for leukemic infiltration of the CSF.
- Treatment and Management ▾: Chemotherapy, targeted therapy and immunotherapy.
*Click ▾ for more information
Introduction
Acute Lymphoblastic Leukemia (ALL) is a highly aggressive hematologic malignancy characterized by the uncontrolled proliferation of immature lymphoid progenitor cells (lymphoblasts) in the bone marrow, peripheral blood, and other extramedullary sites. It is a clonal disorder affecting the early stages of B-cell or T-cell development.
Acute Lymphoblastic Leukemia (ALL) represents a significant clinical challenge with a biphasic incidence: it is the most common childhood cancer, with a peak incidence between ages 2 and 5, yet its prognosis is significantly poorer in adults, particularly those over 60. Therapeutic advances, particularly in pediatric acute lymphoblastic leukemia (ALL), represent one of the major success stories in modern oncology, though optimizing outcomes for adult and relapsed patients remains a critical area of research.
Acute Leukemia
Acute leukemia refers to the rapid proliferation of immature hematopoietic cells (blasts) that fail to differentiate. The fundamental categorization of acute leukemia rests on determining the blast cell's lineage, dividing the disease into lymphoid (acute lymphoblastic leukemia) and myeloid (acute myeloid leukemia) subtypes.
- Acute Lymphoblastic Leukemia (ALL): Originates from lymphoid progenitor cells (lymphoblasts). This lineage gives rise to T-cells and B-cells.
- Acute Myeloid Leukemia (AML): Originates from myeloid progenitor cells (myeloblasts). This lineage gives rise to granulocytes, monocytes, erythrocytes, and platelets.
The distinction between acute lymphoblastic leukemia (ALL) and AML is primarily made through Immunophenotyping (analyzing cell surface markers) and Cytochemistry (examining cytoplasmic components, like Auer rods in AML).
Classification of Acute Lymphoblastic Leukemia (ALL): WHO and Immunophenotyping
The classification of Acute Lymphoblastic Leukemia (ALL) has moved far beyond morphology alone. The modern approach, formalized by the World Health Organization (WHO), integrates three key elements: Lineage, Immunophenotype, and Recurrent Genetic Abnormalities.
WHO Classification: Lineage-Based Grouping
The current WHO classification primarily categorizes acute lymphoblastic leukemia (ALL) based on the lymphoid progenitor cell from which the malignant clone arises, dividing it into two main subtypes.
B-Lymphoblastic Leukemia/Lymphoma (B−ALL) is defined by the malignant transformation of B-cell precursors. It is the dominant subtype, accounting for approximately 80% of childhood acute lymphoblastic leukemia (ALL) cases and 75% of adult cases."
T-Lymphoblastic Leukemia/Lymphoma (T−ALL) is caused by the malignant transformation of T-cell precursors (thymocytes). This subtype, which accounts for 15−20% of all acute lymphoblastic leukemia (ALL) cases, is clinically distinct: it frequently presents with a high White Blood Cell (WBC) count and significant extramedullary disease, most notably a mediastinal mass resulting from blast proliferation in the thymus
Immunophenotyping by Flow Cytometry
Immunophenotyping is the definitive technique used to establish the lineage (B or T) and the precise maturational stage of the leukemic blasts.
Principle of Immunophenotyping
This technique uses flow cytometry to identify specific cell surface or cytoplasmic antigens (markers) expressed by the blasts. Antibodies tagged with fluorescent dyes bind to these markers, allowing thousands of cells to be analyzed rapidly. Learn more about immunophenotyping from our article on ‘Flow Cytometry Immunophenotyping of Blood’.
Key Immunophenotypic Markers for Acute Lymphoblastic Leukemia (ALL)
| Lineage | Markers (Required for Diagnosis) | Additional Key Markers | Developmental Stage (Examples) |
| B-ALL | CD19, Cytoplasmic CD79a, Cytoplasmic CD22 | TdT, CD10 (CALLA), CD20 | Pro-B, Pre-B, Common B-ALL |
| T-ALL | Cytoplasmic CD3 (Most specific T-cell marker) | CD2, CD5, CD7, TdT | Early T-precursor, Cortical T, Mature T-ALL |
*Note on TdT (Terminal Deoxynucleotidyl Transferase): This is an enzyme expressed in the nuclei of both B- and T-lymphoblasts and is a general marker of immaturity in acute leukemia.
WHO Classification Based on Recurrent Genetic Abnormalities
Currently, WHO system includes specific genetic abnormalities as distinct disease entities for acute lymphoblastic leukemia (ALL). Identifying these translocations in acute lymphoblastic leukemia (ALL) is important because they serve as powerful prognostic indicators and can dictate the use of targeted therapy.
A. High-Risk Genetic Subgroups
| Abnormality | WHO Category (Examples) | Significance |
| t(9;22) | BCR−ABL1 fusion (Philadelphia Chromosome) | High-risk; requires Tyrosine Kinase Inhibitor (TKI) therapy (e.g., Imatinib) in addition to chemotherapy. |
| t(4;11) | KMT2A (MLL) rearrangement | High-risk; often seen in infants and associated with early relapse. |
| BCR−ABL1-like ALL | (Ph-like ALL) | Intermediate to high-risk; structurally similar to t(9;22) but is negative for BCR−ABL1. May respond to TKIs. |
B. Favorable/Intermediate-Risk Genetic Subgroups
| Abnormality | WHO Category (Examples) | Significance |
| t(12;21) | ETV6−RUNX1 fusion | Most common translocation in childhood acute lymphoblastic leukemia (ALL); generally associated with a favorable prognosis. |
| Hyperdiploidy | DNA content index >1.16 | Often favorable prognosis, especially with ≥50 chromosomes. |
By combining immunophenotyping and genetic analysis, clinicians can achieve a precise diagnosis, accurately stratify patients into risk groups, and tailor the multi-phase treatment protocol for acute lymphoblastic leukemia (ALL).
Pathogenesis of Acute Lymphoblastic Leukemia (ALL)
The pathophysiology of acute lymphoblastic leukemia (ALL) is rooted in a fundamental disruption of normal lymphoid development, transforming a single, immature progenitor cell into a rapidly proliferating, malignant clone. This process can be divided into three main stages: the genetic initiation, the resulting bone marrow failure, and subsequent extramedullary spread.
The Core Defect: Arrested Differentiation and Uncontrolled Proliferation
Normal hematopoiesis involves a precise, regulated maturation pathway from hematopoietic stem cells to mature lymphocytes. The leukemic process in acute lymphoblastic leukemia (ALL) interrupts this pathway through two mechanisms:
- Arrested Differentiation: A genetic event occurs in a lymphoid progenitor cell (either B-cell or T-cell lineage) that blocks its ability to mature past an early blast stage. This is the Core Defect.
- Uncontrolled Proliferation: Simultaneously, the same genetic abnormality (or a secondary mutation) grants the blast cell independence from normal growth regulation, leading to rapid, exponential cell division.
The combination of an inability to mature and uncontrolled growth leads to the massive accumulation of non-functional, immature lymphoblasts.
Genetic and Molecular Basis: The Oncogenic Drivers
The initiating event is almost always a chromosomal abnormality that results in an oncogenic (cancer-causing) fusion protein. These proteins drive proliferation and inhibit apoptosis (programmed cell death).
BCR−ABL1 Fusion (t(9;22) - Philadelphia Chromosome)
The most clinically significant molecular abnormality for acute lymphoblastic leukemia (ALL) is the translocation of genetic material between chromosome 9 and chromosome 22 fuses the BCR gene with the ABL1 gene. The resulting BCR−ABL1 fusion protein is a constitutively active (always turned "on") tyrosine kinase. This uncontrolled kinase activity signals continuously for cell growth and survival, overriding normal regulatory checks. This mechanism is critical because it offers a direct target for therapy (Tyrosine Kinase Inhibitors).
Other Common Acute Lymphoblastic Leukemia (ALL) Translocations
| Abnormality | Fusion Gene | Clinical Significance |
| t(12;21) | ETV6−RUNX1 | Common in childhood acute lymphoblastic leukemia (ALL), generally associated with a favorable prognosis. |
| t(4;11) | KMT2A−AFF1 (MLL gene) | Associated with very high-risk disease, especially in infants. |
Consequences in the Bone Marrow (Bone Marrow Failure)
As the lymphoblasts rapidly accumulate, the physical space within the bone marrow cavity becomes saturated with malignant cells. The proliferating blasts physically crowd out and suppress the normal hematopoietic stem cells (HSCs) that produce red blood cells, platelets, and mature white blood cells.
This crowding effect leads directly to the classic triad of symptoms resulting from bone marrow failure:
- Anemia: Decreased production of red blood cells (RBCs), leading to fatigue and pallor.
- Thrombocytopenia: Decreased production of platelets, leading to bleeding and bruising.
- Neutropenia: Decreased production of mature neutrophils, leading to increased susceptibility to severe and recurrent infections.
Dissemination and Extramedullary Infiltration
While the bone marrow is the primary site of disease, lymphoblasts circulate in the peripheral blood and have a marked propensity to infiltrate other tissues, a process known as extramedullary disease.
- Central Nervous System (CNS) Infiltration: Blasts can cross the blood-brain barrier and colonize the leptomeninges, leading to CNS involvement. This often requires aggressive targeted treatment (intrathecal chemotherapy) and manifests as headaches, cranial nerve palsies, and vomiting.
- T-ALL Specific Infiltration: Due to its origin from thymocytes, T−ALL commonly presents with a bulky mediastinal mass in the chest.
- Other Sites: The lymphoblasts can also infiltrate the lymph nodes (lymphadenopathy), spleen (splenomegaly), liver (hepatomegaly), and, particularly in males, the testes.
Etiology and Risk Factors
The exact cause of acute lymphoblastic leukemia (ALL) is unknown in the majority of cases, suggesting a complex, multi-step etiology involving both inherited genetic susceptibility and environmental exposures.
Genetic Predisposition (Inherited Factors)
Certain inherited conditions significantly increase the lifetime risk of developing acute lymphoblastic leukemia (ALL), particularly B-ALL, by impairing repair mechanisms or affecting hematopoietic stability.
Congenital Syndromes
| Syndrome | Associated Mechanism/Features | Increased Risk of ALL |
| Down Syndrome (Trisomy 21) | Presence of an extra chromosome 21. | 10−20 times higher risk, especially before age 5. |
| Klinefelter Syndrome | XXY sex chromosomes. | Increased risk of B-ALL. |
| Neurofibromatosis Type 1 (NF1) | Affects the RAS signaling pathway, leading to uncontrolled proliferation. | Increased risk of various malignancies, including ALL. |
| Li-Fraumeni Syndrome | Mutation in the tumor suppressor gene p53. | Predisposes to multiple cancers, including leukemia. |
| Fanconi Anemia | Defective DNA repair. | Primarily associated with AML, but also increased risk of ALL. |
Inherited Immune Deficiency Disorders
Conditions that lead to chronic immune dysfunction or dysregulated lymphocyte production can also increase risk.
- Ataxia-telangiectasia: Defect in ATM gene (DNA repair), leading to T-cell defects and increased risk of T−ALL.
- Wiskott-Aldrich Syndrome: T-cell and B-cell functional defects.
Environmental Exposures
While less defined than for acute myeloid leukemia (AML), several environmental factors are implicated in the development of acute lymphoblastic leukemia (ALL).
Ionizing Radiation
Exposure to high-dose ionizing radiation (e.g., atomic bomb survivors, high-dose medical radiation) is a clear risk factor for various leukemias, including acute lymphoblastic leukemia (ALL). However, prenatal X-ray exposure is thought to be a minor risk, and there is no proven link to background radiation levels.
Chemical and Drug Exposure
- Chemotherapeutic Agents: Prior exposure to certain cytotoxic chemotherapy drugs (particularly Topoisomerase II inhibitors like Etoposide) for other cancers is a known cause of secondary leukemias, though more commonly AML.
- Pesticides/Herbicides: Exposure to certain household and agricultural chemicals has been epidemiologically linked to a slightly increased risk of childhood acute lymphoblastic leukemia (ALL), though the evidence remains debated.
- Benzene: A known human carcinogen strongly associated with AML, its link to acute lymphoblastic leukemia (ALL) is less definitive but remains a suspected risk factor.
The Role of Infections and the "Delayed Infection" Hypothesis
This hypothesis is a key model used to explain the etiology of the largest group of acute lymphoblastic leukemia (ALL) cases: childhood B-ALL in otherwise healthy individuals.
The Theory (Proposed by Mel Greaves)
Childhood acute lymphoblastic leukemia (ALL) is a two-step process:
- First Hit (Prenatal): A silent, pre-leukemic genetic lesion (e.g., the ETV6−RUNX1 fusion) occurs in utero. This is common and does not cause leukemia on its own.
- Second Hit (Postnatal): The child with the pre-leukemic clone experiences an uncommon or delayed exposure to common infections during the first few years of life.
The delayed exposure triggers an abnormally intense and dysregulated immune response in a susceptible child. This chronic, prolonged immune stimulation drives the proliferation of the pre-leukemic cells, causing them to acquire the second necessary mutation, thus initiating overt acute lymphoblastic leukemia (ALL).
This hypothesis is supported by data showing higher acute lymphoblastic leukemia (ALL) incidence in populations with smaller social networks, less early social mixing (less exposure to common infections early in life), and rapid population mixing.
Clinical Presentation of Acute Lymphoblastic Leukemia (ALL)
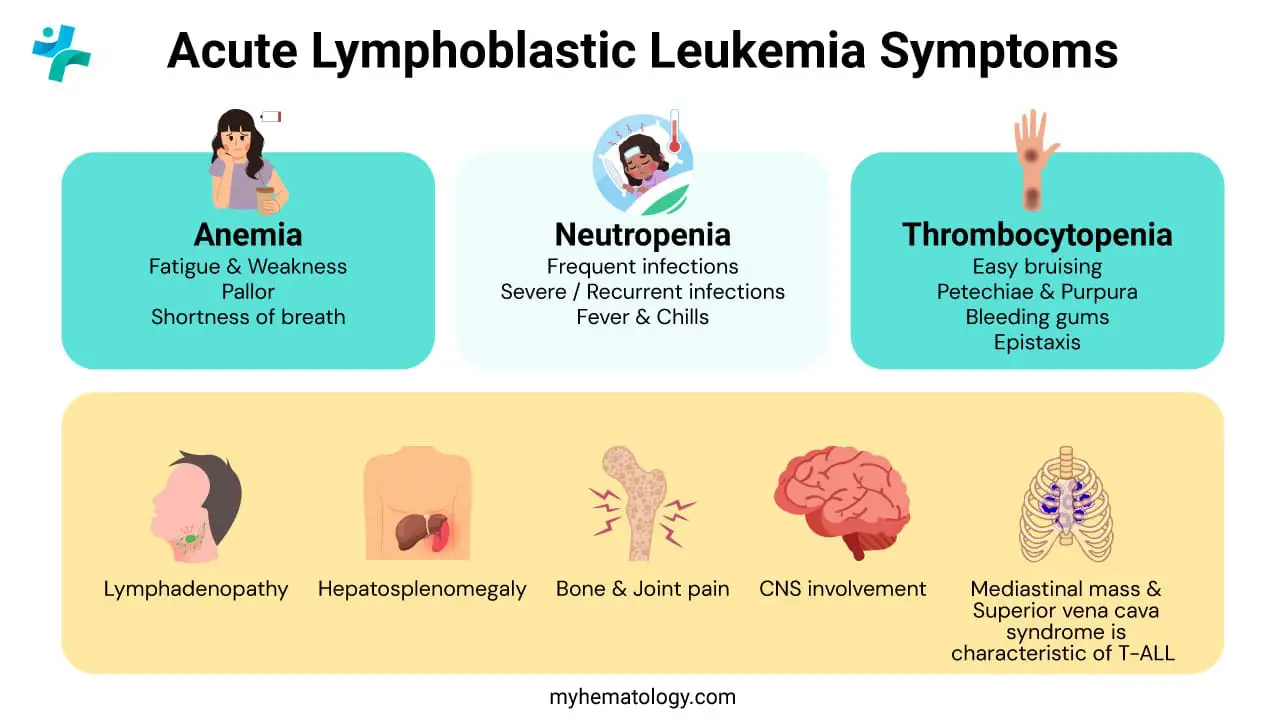
The clinical presentation of acute lymphoblastic leukemia (ALL) is often acute and non-specific, leading to delayed diagnosis. Symptoms arise directly from the consequences of bone marrow failure and the infiltration of organs by lymphoblasts.
Signs and Symptoms of Bone Marrow Failure (Cytopenias)
The rapid proliferation of lymphoblasts in the bone marrow physically suppresses the production of normal hematopoietic cells, resulting in a reduction of circulating mature red cells, white cells, and platelets (cytopenias).
Anemia (Decreased Red Blood Cells)
This is due to suppression of erythropoiesis by crowding lymphoblasts.
- Fatigue and Weakness: The most common initial complaints, often slowly progressive.
- Pallor: Paleness of the skin and conjunctiva.
- Dyspnea (Shortness of Breath): Especially on exertion, due to decreased oxygen-carrying capacity.
Thrombocytopenia (Decreased Platelets)
Suppression of megakaryopoiesis leads to signs of abnormal bleeding due to impaired clot formation.
- Petechiae and Purpura: Pinpoint hemorrhages (petechiae) or larger blotches (purpura) are common, often appearing spontaneously.
- Easy Bruising (Ecchymoses).
- Mucosal Bleeding: Epistaxis (nosebleeds) and gingival (gum) bleeding.
Neutropenia (Decreased Mature Neutrophils)
This is due to suppression of granulopoiesis.
- Infection and Fever: Recurrent, persistent, or unusually severe bacterial or fungal infections.
- Fever of Unknown Origin (FUO): A persistent, unexplained fever is a classic presentation, demanding immediate evaluation for neutropenic sepsis (an oncologic emergency).
Extramedullary Involvement (Organ Infiltration)
Lymphoblasts circulate and infiltrate non-hematopoietic organs, leading to mass effects or organ dysfunction.
Organomegaly and Lymphadenopathy
- Lymphadenopathy: Enlarged, non-tender, firm lymph nodes (neck, axillae, groin).
- Hepatosplenomegaly: Enlargement of the liver and/or spleen due to blast infiltration, often palpable on physical exam.
Bone and Joint Pain
Infiltration of the marrow cavity causes expansion of the bony cortex and periosteal irritation. This leads to deep, aching bone pain, most common in the long bones or vertebrae. In children, this may present as refusal to bear weight or walk, sometimes mimicking juvenile idiopathic arthritis.
Central Nervous System (CNS) Involvement
Blasts cross the blood-brain barrier and infiltrate the leptomeninges (layers covering the brain and spinal cord). This leads to manifestations of increased intracranial pressure causing:
- Persistent Headaches and Vomiting.
- Cranial Nerve Palsies: Particularly of the facial (CN VII) or abducens (CN VI) nerves.
T-ALL Specific Findings
Mediastinal Mass is highly characteristic of T−ALL due to its origin from thymocytes in the thymus. The mass can compress adjacent structures, leading to life-threatening Superior Vena Cava (SVC) Syndrome (facial plethora, neck and upper chest vein distention, shortness of breath).
Other Sites
- Testicular Infiltration: Unilateral or bilateral painless, firm testicular swelling (a known "sanctuary site" for leukemic cells).
- Skin: Leukemic infiltration of the skin (leukemia cutis), though less common than in AML.
Laboratory Investigations and Diagnosis of Acute Lymphoblastic Leukemia
The diagnosis of acute lymphoblastic leukemia (ALL) is a multi-step process that confirms the presence of lymphoblasts, determines the specific WHO subtype, and identifies risk-stratifying genetic abnormalities.
Initial Screening
Complete Blood Count (CBC) and Differential
Typically shows pancytopenia (anemia, thrombocytopenia, and neutropenia) due to bone marrow suppression.
- White Blood Cell (WBC) Count: Highly variable. The total WBC count can be low, normal, or extremely high (often >100×109/L), but the common feature is the presence of blasts.
Peripheral Blood Smear
Visualization of lymphoblasts (often >20%). These are large, immature cells with scant cytoplasm, a high nucleus-to-cytoplasm ratio, and nucleoli.
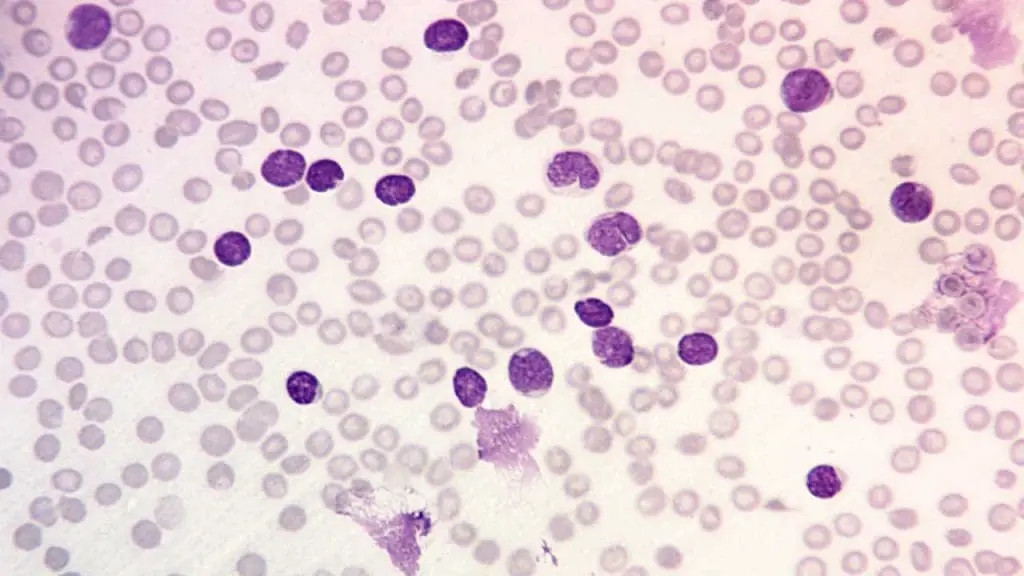
Definitive Diagnosis: Bone Marrow Biopsy
A bone marrow aspirate and biopsy are essential to confirm the diagnosis and provide material for detailed subtyping. Diagnosis requires the presence of ≥20% lymphoblasts in the bone marrow aspirate (or peripheral blood if blood is circulating). Historically, the morphology is classified by the FAB system (L1,L2,L3), although this is largely superseded by immunophenotyping.
Classification and Risk Stratification
These studies are performed on bone marrow or blood to classify the acute lymphoblastic leukemia (ALL) subtype and identify key prognostic markers.
Immunophenotyping (Flow Cytometry)
This investigation determines the exact lineage (B-cell vs. T-cell) and maturational stage by identifying cluster of differentiation (CD) surface markers. This is the foundation of the WHO classification.
- B-ALL Markers: Positive for CD19, CD20, CD22, CD79a, and PAX5.
- T-ALL Markers: Positive for CD2, CD3 (cytoplasmic or surface), CD5, CD7, and often CD4/CD8.
Cytogenetics and Molecular Genetics
These investigations identify recurrent chromosomal translocations and genetic mutations that are crucial for risk stratification and selecting targeted therapy.
- Fluorescence In Situ Hybridization (FISH): Used to rapidly detect key translocations like t(9;22) (BCR−ABL1).
- Polymerase Chain Reaction (PCR)/Next-Generation Sequencing (NGS): Used to detect fusion genes (e.g., BCR−ABL1, ETV6−RUNX1) and other prognostic mutations (e.g., IKZF1 deletions).
CNS Assessment
Lumbar Puncture (LP) is mandatory at diagnosis (unless contraindicated due to high WBC or thrombocytopenia) to check for central nervous system (CNS) involvement. The cerebrospinal fluid (CSF) is analyzed for the presence of blasts. If blasts are found, it upstages the disease and requires intensified intrathecal therapy.
Pre-Treatment Staging and Labs
- Metabolic Panel: Checks for tumor lysis syndrome (TLS) risk (high uric acid, potassium, phosphate, and low calcium) and assesses kidney and liver function.
- Coagulation Studies: Important due to potential for severe thrombocytopenia and consumption coagulopathy.
- Infectious Disease Screening: Testing for Hepatitis B and C, HIV, and CMV is essential as these infections can reactivate during immunosuppressive chemotherapy.
Treatment and Management of ALL
The treatment of acute lymphoblastic leukemia (ALL) is highly complex, prolonged (typically 2 to 3 years), and risk-adapted, meaning the intensity of therapy is tailored based on patient age, WBC count, genetic markers, and response to initial treatment (Minimal Residual Disease or MRD).
Chemotherapy Phases (The Three Pillars)
Chemotherapy remains the cornerstone of acute lymphoblastic leukemia (ALL) treatment and is generally divided into three sequential phases.
Induction Therapy
- Goal: To rapidly induce a Complete Remission (CR), defined as the disappearance of blasts from the peripheral blood, reduction of blasts in the bone marrow to <5%, and recovery of normal blood counts.
- Duration: Approximately 4 weeks.
- Drugs: Intensive multi-agent regimen, typically including Vincristine, a corticosteroid (Prednisone or Dexamethasone), and an anthracycline (Daunorubicin or Doxorubicin), often combined with L-asparaginase.
- Prognostic Marker: Minimal Residual Disease (MRD) status measured at the end of induction is the single most powerful predictor of relapse risk.
Consolidation/Intensification Therapy
- Goal: To eliminate any remaining subclinical disease (known as MRD) that survived the induction phase.
- Duration: Several months.
- Drugs: Rotating courses of non-cross-resistant drugs (Methotrexate, Cytarabine, Cyclophosphamide) to prevent the development of drug resistance.
Maintenance Therapy
- Goal: To sustain remission and eliminate any late-cycling leukemic cells.
- Duration: Usually 2 to 3 years.
- Drugs: Low-intensity daily oral 6−Mercaptopurine (6−MP) and weekly oral Methotrexate, combined with intermittent pulses of Vincristine and corticosteroids.
Central Nervous System (CNS) Prophylaxis
Since acute lymphoblastic leukemia (ALL) cells frequently invade the CNS ("sanctuary site"), prophylaxis is mandatory for all patients throughout all phases of treatment.
- Intrathecal Chemotherapy: Direct injection of drugs (typically Methotrexate, Cytarabine, or corticosteroids) into the cerebrospinal fluid (CSF) via a lumbar puncture (LP).
- High-dose Systemic Chemotherapy: Drugs like Methotrexate and Cytarabine that can cross the blood-brain barrier at high concentrations.
Targeted and Immunotherapies
These treatments target specific genetic abnormalities or utilize the patient's immune system.
Tyrosine Kinase Inhibitors (TKIs)
For BCR−ABL1-positive acute lymphoblastic leukemia (ALL) (Philadelphia chromosome, t(9;22)).
- Drugs: Imatinib, Dasatinib, or Nilotinib are added to standard chemotherapy. This dramatically improved outcomes for this high-risk group.
Monoclonal Antibodies (mAbs)
- Blinatumomab: A CD19/CD3 bispecific T-cell engager (BiTE) that connects T-cells to B-cells, leading to B-cell destruction. Used for MRD-positive and relapsed/refractory ALL.
- Inotuzumab Ozogamicin: An antibody-drug conjugate targeting CD22.
CAR T-cell Therapy (Chimeric Antigen Receptor T-cells)
The patient's own T-cells are genetically modified ex vivo to express a CAR that recognizes a leukemia-specific antigen, most commonly CD19. These modified T-cells are then infused back into the patient to hunt and destroy the malignant cells. It is highly effective for relapsed or refractory B−ALL in both children and adults.
Hematopoietic Stem Cell Transplantation (HSCT)
It is primarily used for patients with high-risk ALL (based on genetics, age, or poor MRD response) or those who experience relapse. The goal is to replace the patient's faulty hematopoietic system with a healthy one, and to benefit from the graft-versus-leukemia (GVL) effect, where donor immune cells recognize and kill residual leukemic cells.
Supportive Care
Aggressive management of treatment-related toxicities is crucial for survival.
- Tumor Lysis Syndrome (TLS): Prevention with intravenous hydration and medications (Allopurinol or Rasburicase) to manage high uric acid levels.
- Infection Prophylaxis and Treatment: Due to prolonged neutropenia, prophylactic antibiotics, antivirals, and antifungals are used. Prompt, broad-spectrum antibiotic administration is mandatory for any patient with fever and neutropenia.
- Blood Component Support: Transfusions of packed red blood cells and platelets as needed.
Management of Relapsed and Refractory Disease
Relapse is a major challenge, often occurring in the bone marrow, CNS, or testicles. The goal of therapy for relapsed disease is to achieve a second, deep remission, ideally MRD-negative, to bridge the patient to allo-HSCT or CAR T-cell therapy. Novel agents (Blinatumomab, Inotuzumab) have significantly improved outcomes in this setting.
Prognosis and Follow-Up
The prognosis for acute lymphoblastic leukemia (ALL) has improved dramatically, particularly in pediatric cases, with cure rates exceeding 90% in many pediatric protocols. However, outcomes remain highly variable based on specific disease characteristics and therapeutic response.
Prognostic Factors
The intensity of acute lymphoblastic leukemia (ALL) therapy is always risk-adapted, meaning it is tailored based on factors that predict the likelihood of relapse.
Patient-Specific Factors
- Age: Best outcomes are seen in children aged 1 to 9 years. Infants (<1 year) and adults (>50 years) generally have a poorer prognosis due to a higher incidence of adverse genetic subtypes.
- Initial White Blood Cell (WBC) Count: A low WBC count (<50×109/L at diagnosis) is typically favorable, while a very high WBC count suggests a higher tumor burden and is often a high-risk feature.
Genetic and Cytogenetic Factors (The WHO Subtypes)
- Favorable: The t(12;21) (ETV6−RUNX1) translocation is associated with the best long-term outcomes. Hyperdiploidy (more than 50 chromosomes) is also considered favorable.
- Unfavorable (High-Risk):
- t(9;22) (BCR−ABL1 or Philadelphia Chromosome) requires the addition of targeted Tyrosine Kinase Inhibitors (TKIs).
- KMT2A gene rearrangements (e.g., t(4;11)) are highly adverse, especially in infants.
- T-ALL generally has a worse prognosis than B-ALL but has improved significantly with modern, intensified protocols.
Response to Therapy: Minimal Residual Disease (MRD)
MRD is the detection of small numbers of leukemic cells (often <0.01%) that remain after treatment and are undetectable by routine microscopy. It is assessed using highly sensitive techniques like flow cytometry or PCR.
MRD status is the single most powerful prognostic factor.
- MRD-Negativity: Patients who achieve this status after induction and consolidation have the lowest risk of relapse.
- Persistent MRD: Suggests resistance to chemotherapy and necessitates treatment intensification, often including targeted therapy or Hematopoietic Stem Cell Transplantation (HSCT).
Long-Term Follow-up (Survivorship Care)
Given the high cure rates, particularly in children, focus shifts to managing the potential long-term complications of intensive chemotherapy and radiation.
Monitoring for Relapse
The highest risk of relapse is within the first 2 years after diagnosis. Surveillance continues after the completion of maintenance therapy, with regular CBCs and physical exams, especially monitoring for signs of extramedullary relapse (CNS, testes).
Late Effects of Therapy
Survivors of acute lymphoblastic leukemia (ALL) are at risk for life-long complications resulting from the curative treatment.
- Cardiotoxicity: Damage to the heart muscle from cumulative doses of anthracyclines (e.g., Daunorubicin), requiring lifelong surveillance.
- Neurocognitive Deficits: Issues with attention, memory, and executive function, especially common in patients who received CNS radiation.
- Endocrine Dysfunction: Thyroid dysfunction, growth failure, and reduced fertility potential are common and require hormone replacement therapy or monitoring.
- Musculoskeletal Issues: Osteoporosis and avascular necrosis (osteonecrosis) due to high-dose steroid therapy.
- Secondary Malignancies: An elevated risk of developing other cancers later in life (e.g., Acute Myeloid Leukemia (AML)).
Survivorship Clinics
Acute lymphoblastic leukemia (ALL) patients are often transitioned to specialized survivorship clinics that provide:
- A written summary of their diagnosis and treatment.
- Guidelines for necessary health screenings (e.g., cardiac imaging, bone density scans).
- Counseling regarding potential late effects and healthy lifestyle choices.
Frequently Asked Questions (FAQs)
Why is Acute Lymphoblastic Leukemia (ALL) treatment much longer than treatment for Acute Myeloid Leukemia (AML)?
Acute lymphoblastic leukemia (ALL) treatment is typically long (2–3 years) because of the maintenance phase, which is crucial to preventing relapse. Leukemic lymphoblasts are prone to dormancy and are highly susceptible to chemotherapy at the low doses used in maintenance. AML, conversely, is usually treated with several intense cycles of consolidation chemotherapy over 6–12 months, without a prolonged maintenance phase.
What is the main clinical difference between B-ALL and T-ALL presentation?
T-ALL has a higher propensity for extramedullary disease, frequently presenting with a mediastinal mass (often causing respiratory symptoms) and a higher risk of CNS involvement at diagnosis. B-ALL typically presents primarily with symptoms related to bone marrow failure (fatigue, easy bruising, infection).
What is the significance of the Philadelphia-like ALL (Ph-like ALL) subtype?
Ph-like ALL is a molecularly high-risk subtype that lacks the BCR-ABL1 fusion but shares a similar gene expression profile. Crucially, its genetic alterations (e.g., JAK mutations, CRLF2 rearrangements) often activate similar signaling pathways, making it potentially targetable with TKIs or JAK inhibitors, improving prognosis when identified early.
What is the main toxicity concern with CAR T-cell therapy?
The two most significant acute toxicities are Cytokine Release Syndrome (CRS) and Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS). CRS is a systemic inflammatory response managed with supportive care and the IL-6 receptor antagonist, tocilizumab. ICANS involves a range of neurological symptoms.
Glossary of Medical Terms
- Lymphoblast: The immature, malignant lymphoid progenitor cell that is the characteristic cell of ALL.
- Cytogenetics: The study of chromosome structure and number within cells, crucial for identifying prognostic translocations like t(9;22) (Philadelphia Chromosome).
- Flow Cytometry: A high-speed laboratory technique used for immunophenotyping; it identifies cells based on the presence of specific surface or intracellular protein markers (e.g., CD19, CD3) to determine the leukemic cell lineage.
- Minimal Residual Disease (MRD): The presence of a small number of remaining leukemic cells after treatment, detectable only by highly sensitive techniques (e.g., PCR, deep sequencing); it is the strongest prognostic factor.
- BCR-ABL1: A fusion gene resulting from the t(9;22) translocation (Philadelphia Chromosome), which produces a constitutively active tyrosine kinase.
- Tyrosine Kinase Inhibitor (TKI): A class of drugs (e.g., Imatinib, Dasatinib) that specifically blocks the activity of tyrosine kinase enzymes, essential for treating Ph+ ALL.
- Intrathecal Chemotherapy: The injection of chemotherapy drugs (most commonly Methotrexate) directly into the cerebrospinal fluid (CSF) via a lumbar puncture to treat or prevent central nervous system (CNS) leukemia.
- Bispecific T-cell Engager (BiTE): An engineered monoclonal antibody (e.g., Blinatumomab) that simultaneously binds a target on the cancer cell (CD19) and a marker on the patient's T-cell (CD3), effectively linking the two to induce tumor cell lysis.
- Antibody-Drug Conjugate (ADC): A targeted therapy (e.g., Inotuzumab Ozogamicin) that links a cytotoxic payload (chemotherapy) to an antibody that recognizes a specific antigen (CD22) on the cancer cell, allowing for targeted drug delivery.
- CAR T-cell Therapy (Chimeric Antigen Receptor T-cell Therapy): A sophisticated form of immunotherapy where a patient's own T-cells are genetically modified ex vivo to express a CAR that recognizes a specific tumor antigen (CD19), expanded, and re-infused.
- Allogeneic HSCT (allo-HSCT): Allogeneic Hematopoietic Stem Cell Transplantation; the infusion of stem cells from a healthy donor to replace the patient's cancerous bone marrow and reconstitute their immune system.
Disclaimer: This article is intended for informational purposes only and is specifically targeted towards medical students. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Puckett Y, Chan O. Acute Lymphocytic Leukemia. [Updated 2023 Aug 26]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK459149/
- https://www.cancer.gov/types/leukemia/hp/adult-all-treatment-pdq
- https://www.cancer.gov/types/leukemia/hp/child-all-treatment-pdq
- Terwilliger, T., Abdul-Hay, M. Acute lymphoblastic leukemia: a comprehensive review and 2017 update. Blood Cancer J. 7, e577 (2017). https://doi.org/10.1038/bcj.2017.53
- Elgazar, S., & Constantinou, C. (2024). Paediatric Acute Lymphoblastic Leukaemia: A Narrative Review of Current Knowledge and Advancements. Current oncology reports, 26(12), 1586–1599. https://doi.org/10.1007/s11912-024-01608-4
- Bloom, M., Maciaszek, J. L., Clark, M. E., Pui, C. H., & Nichols, K. E. (2020). Recent advances in genetic predisposition to pediatric acute lymphoblastic leukemia. Expert review of hematology, 13(1), 55–70. https://doi.org/10.1080/17474086.2020.1685866
- Fielding, A. K., & Goldstone, A. H. (2020). Acute lymphoblastic leukaemia (ALL) things come to those who wait: 60 years of progress in the treatment of adult ALL. British journal of haematology, 191(4), 558–561. https://doi.org/10.1111/bjh.17166
- Chen, Z., Xin, Q., Wei, W., & Wu, Y. (2023). The pathogenesis and development of targeted drugs in acute T lymphoblastic leukaemia. British journal of pharmacology, 180(8), 1017–1037. https://doi.org/10.1111/bph.16029