Key Takeaways
Glanzmann thrombasthenia is a rare, inherited bleeding disorder characterized by defective or deficient platelet αIIbβ3 (GPIIb/IIIa) integrin leading impaired platelet aggregation despite normal platelet count.
Pathophysiology ▾: αIIbβ3 is crucial for platelet-platelet binding via fibrinogen and vWF. The deficiency/dysfunction of this protein prevents stable platelet plug formation. Initial platelet adhesion may be normal, but aggregation is severely impaired.
Causes ▾: Mutations in ITGA2B (encoding αIIb) or ITGB3 (encoding β3) genes inherited in an autosomal recessive inheritance pattern.
- Primarily mucocutaneous bleeding (nosebleeds, gum bleeding, bruising, petechiae).
- Heavy menstrual bleeding (menorrhagia) in females.
- Prolonged bleeding after minor injuries, surgery, or dental procedures.
- Less frequent but serious bleeds (GI, intracranial, joint).
- Onset typically in infancy or early childhood.
- Normal platelet count and morphology.
- Prolonged bleeding time/PFA-100.
- Absent or severely reduced platelet aggregation to all agonists EXCEPT ristocetin (key finding).
- Reduced or absent αIIbβ3 expression by flow cytometry.
- Poor or absent clot retraction.
- Genetic testing confirms mutations in ITGA2B or ITGB3.
- Acute Bleeding: Local hemostatic measures, platelet transfusions (primary), recombinant activated factor VIIa (rFVIIa).
- Prophylaxis: Avoid antiplatelet drugs, caution with procedures, antifibrinolytics for mucosal bleeding.
- Alloimmunization Management: HLA-matched platelets, rFVIIa, immunosuppression.
- Supportive care, patient education, and genetic counseling are crucial.
*Click ▾ for more information
Introduction
Glanzmann thrombasthenia (GT) is a rare, inherited bleeding disorder characterized by a qualitative or quantitative deficiency of the platelet integrin αIIbβ3 (also known as glycoprotein IIb/IIIa or GPIIb/IIIa). This deficiency impairs the ability of platelets to aggregate, which is a crucial step in forming a blood clot to stop bleeding.
Pathophysiology of Glanzmann Thromboasthenia
At its core, the pathophysiology of Glanzmann thrombasthenia revolves around a defect in platelet aggregation, the process by which platelets stick to each other to form a stable plug at the site of vascular injury. This defect stems directly from the impaired function or absence of the αIIbβ3 (GPIIb/IIIa) integrin on the surface of platelets.
Normal Platelet Plug Formation
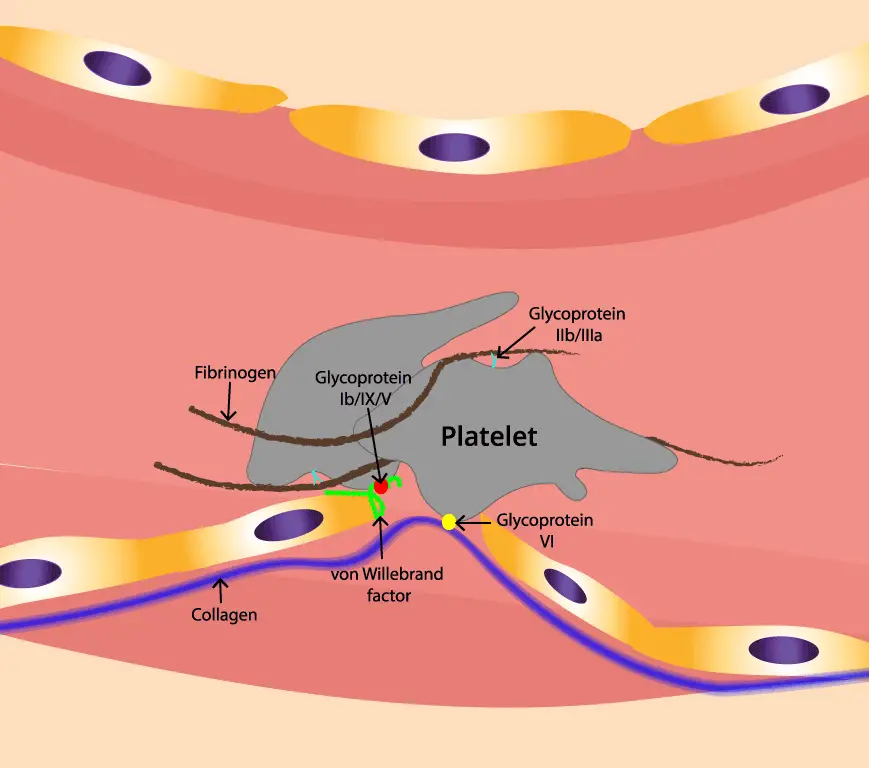
When a blood vessel is injured, the underlying collagen and von Willebrand factor (vWF) are exposed. Platelets in the bloodstream adhere to these exposed subendothelial components via specific receptors, primarily the GPIb-IX-V complex binding to vWF. This initial adhesion slows down platelets at the injury site.
The initial adhesion triggers platelet activation, leading to a conformational change in αIIbβ3, increasing its affinity for its ligands. Activated platelets also release various substances from their granules, such as ADP and thromboxane A2, which further activate nearby platelets.
The activated αIIbβ3 integrins on the surface of adjacent platelets bind to fibrinogen (a soluble plasma protein) and, to a lesser extent, to von Willebrand factor (vWF). Fibrinogen acts as a bridge, with each molecule binding to αIIbβ3 on two different platelets, effectively cross-linking them and forming a platelet plug. This aggregation is crucial for hemostasis (stopping bleeding).
The initial platelet plug is then stabilized by the coagulation cascade, leading to the generation of thrombin, which converts fibrinogen into fibrin. Fibrin forms a meshwork that reinforces the platelet plug, creating a stable blood clot. Platelet contraction, mediated by αIIbβ3 binding to fibrin, further strengthens the clot.
Impaired αIIbβ3 Function in Glanzmann Thrombasthenia
In Glanzmann thrombasthenia, the fundamental problem lies in the qualitative or quantitative abnormality of the αIIbβ3 integrin.
- Quantitative Deficiency: There is a significantly reduced number or even complete absence of αIIbβ3 receptors on the platelet surface. This is characteristic of Type I Glanzmann Thrombasthenia.
- Qualitative Abnormality: The αIIbβ3 receptors are present in normal or near-normal numbers, but they are structurally or functionally defective and cannot bind effectively to their ligands (primarily fibrinogen and vWF). This is characteristic of Type II Glanzmann Thrombasthenia. There's also a rarer variant form where the receptor might have some residual function.
The αIIbβ3 integrin, also known as Glycoprotein IIb/IIIa (GPIIb/IIIa), is a crucial transmembrane protein found in high abundance on the surface of platelets and their precursor cells, megakaryocytes.
The primary function of αIIbβ3 is to mediate platelet aggregation, a critical step in the formation of a hemostatic plug to stop bleeding. This function is achieved through its ability to bind to specific adhesive ligands, most importantly fibrinogen and, under high shear stress conditions, von Willebrand factor (vWF). It can also bind to other ligands like fibronectin, vitronectin, and thrombospondin, which can modulate platelet aggregation and adhesion.
The impaired function of αIIbβ3 directly disrupts the process of platelet aggregation. Without functional αIIbβ3 receptors that can bind fibrinogen and vWF effectively, platelets cannot adhere to each other to form a stable platelet plug. Even though initial platelet adhesion to the injured vessel wall (mediated by GPIb-IX-V) may be normal, the subsequent crucial step of platelet aggregation is severely compromised.
The inability to form a robust platelet plug leads to prolonged bleeding, especially from mucocutaneous sites where initial hemostasis relies heavily on platelet aggregation.
Genetic Basis and Causes of Glanzmann Thrombasthenia
Glanzmann thrombasthenia (GT) is an inherited disorder with an autosomal recessive inheritance pattern. To develop Glanzmann thrombasthenia, an individual must inherit two copies of a mutated gene, one from each parent.
Glanzmann thrombasthenia is caused by mutations in one of two genes located on chromosome 17:
- ITGA2B gene: This gene provides the instructions for making the αIIb subunit of the αIIbβ3 integrin.
- ITGB3 gene: This gene provides the instructions for making the β3 subunit of the αIIbβ3 integrin.
Both subunits are essential for the formation of a functional αIIbβ3 receptor on the surface of platelets. Mutations in either of these genes can lead to Glanzmann thrombasthenia.
Various types of mutations in these genes can occur, including missense mutations (single amino acid changes), nonsense mutations (premature stop codons), deletions, and insertions. These mutations can lead to the reduced expression or the production of non-functional αIIbβ3 protein.
Correlation Between Genotype and Phenotype
While there is significant variability in the severity of bleeding symptoms among individuals with Glanzmann thrombasthenia, there is some correlation between the type and location of the genetic mutation and the amount of functional αIIbβ3 protein produced.
- Type I Glanzmann Thrombasthenia: Usually associated with mutations that lead to a severe reduction (less than 5% of normal) or complete absence of αIIbβ3 on the platelet surface. These patients often experience more severe bleeding.
- Type II Glanzmann Thrombasthenia: Typically results from mutations that allow for the expression of a reduced amount of αIIbβ3 (5-20% of normal) or the expression of a dysfunctional receptor. These patients may have milder bleeding symptoms compared to Type I.
- Variant Glanzmann Thrombasthenia: In this rare form, the amount of αIIbβ3 on the platelet surface may be near normal, but the receptor is non-functional due to specific mutations affecting its ability to bind to ligands.
- Acquired Glanzmann Thrombasthenia (Rare): In very rare cases, individuals can develop an acquired form of Glanzmann thrombasthenia later in life. This occurs when the body produces autoantibodies that target and block the function of the αIIbβ3 integrin on platelets. This condition is not inherited and can be associated with certain underlying hematologic or autoimmune disorders.
Signs and Symptoms of Glanzmann Thrombasthenia
The signs and symptoms of Glanzmann thrombasthenia (GT) are primarily related to an increased tendency to bleed due to the impaired ability of platelets to form a stable plug. The severity and frequency of bleeding episodes can vary significantly among affected individuals, even within the same family. However, certain patterns are commonly observed.
Mucocutaneous Bleeding (Bleeding from Mucous Membranes and Skin)

This is the most common category of bleeding in Glanzmann thrombasthenia and often presents early in life.
- Epistaxis (Nosebleeds): Frequent and sometimes severe nosebleeds are a hallmark symptom. They can be spontaneous or triggered by minor trauma.
- Gingival Bleeding (Gum Bleeding): Bleeding from the gums, especially after brushing teeth or dental procedures, is very common.
- Bruising (Ecchymoses): Individuals with Glanzmann thrombasthenia tend to bruise easily and extensively, often with minimal or no apparent injury. The bruises can range in size and may take longer than usual to resolve.
- Petechiae: These are small, pinpoint red or purple spots on the skin, resulting from superficial capillary bleeding. They can occur in crops, particularly on the extremities.
- Prolonged Bleeding from Minor Cuts and Abrasions: Even small cuts or scrapes can bleed for a longer duration than in individuals with normal platelet function.
Bleeding in Females
- Menorrhagia (Heavy Menstrual Bleeding): This is a significant issue for females with Glanzmann thrombasthenia and can lead to anemia and a reduced quality of life. Menstrual periods can be excessively heavy and prolonged, often requiring medical intervention.
Post-Traumatic and Post-Surgical Bleeding
- Prolonged Bleeding After Surgery: Any surgical procedure, even minor ones, carries a risk of prolonged and excessive bleeding in individuals with GT. This necessitates careful planning and management.
- Bleeding After Dental Procedures: Tooth extractions and other dental work can lead to significant and prolonged bleeding from the gums and extraction sites.
- Bleeding After Trauma: Even minor injuries can result in more bleeding than expected. Major trauma can lead to severe and life-threatening hemorrhage.
Less Frequent but Potentially Serious Bleeding
- Gastrointestinal Bleeding: Bleeding from the stomach or intestines can occur, leading to symptoms like black, tarry stools (melena) or vomiting blood (hematemesis). This can sometimes be significant and require medical attention.
- Hematuria (Blood in Urine): Bleeding from the kidneys or urinary tract can result in blood in the urine.
- Intracranial Hemorrhage: This is a rare but very serious complication that involves bleeding within the skull. It can present with severe headache, neurological deficits, and can be life-threatening.
- Joint Bleeds (Hemarthrosis): While less common than in hemophilia, joint bleeding can occur in individuals with GT, especially after significant trauma. This can lead to pain, swelling, and long-term joint damage if not managed appropriately.
Onset of Symptoms
Symptoms of Glanzmann thrombasthenia typically manifest early in life, often in infancy or early childhood, with easy bruising and mucocutaneous bleeding episodes. Menorrhagia usually becomes apparent at the onset of menstruation in females.
Laboratory Investigations in Glanzmann Thromboasthenia
Laboratory investigations are crucial for diagnosing Glanzmann thrombasthenia (GT). Since Glanzmann thrombasthenia is characterized by dysfunctional platelets despite a normal count, standard coagulation tests are usually normal. The focus of laboratory testing is on assessing platelet function and identifying the deficiency or abnormality of the αIIbβ3 integrin.
Initial Screening Tests
- Complete Blood Count (CBC) and Peripheral Blood Smear: Platelet count is typically normal. Platelet morphology on the peripheral smear is also usually normal in terms of size and appearance. In rare cases, some mutations might be associated with platelets of unequal size. The red blood cell count may be low in patients with chronic bleeding and iron deficiency.
- Prothrombin Time (PT) and Activated Partial Thromboplastin Time (aPTT): These tests assess the coagulation cascade (clotting factors) and are usually normal in Glanzmann thrombasthenia, as the defect lies in platelet function, not the coagulation pathway.
- Bleeding Time (BT): The bleeding time is typically prolonged, reflecting the impaired ability of platelets to form an effective primary hemostatic plug. However, this test is less standardized and its use has decreased in many centers.
- Platelet Function Analyzer (PFA-100): This automated test measures the time it takes for platelets to plug a small aperture coated with collagen and epinephrine or ADP under flow conditions. In Glanzmann thrombasthenia, the closure time is usually prolonged or not measurable, indicating impaired platelet function.
Specific Platelet Function Tests
- Light Transmission Aggregometry (LTA): This is considered the gold standard for diagnosing Glanzmann thrombasthenia. It measures the change in light transmission through a platelet-rich plasma sample after the addition of various platelet agonists. In Glanzmann thrombasthenia, there is absent or severely reduced platelet aggregation in response to all physiological agonists that primarily act through αIIbβ3, such as:
- Adenosine diphosphate (ADP)
- Collagen
- Epinephrine
- Thrombin
- Arachidonic acid
Characteristic Finding: Platelet aggregation in response to ristocetin is typically normal or near normal. Ristocetin induces platelet aggregation by binding to von Willebrand factor (vWF) and GPIb/IX/V on platelets, bypassing the need for functional αIIbβ3 for the initial agglutination (though αIIbβ3 may play a minor role at very high concentrations of ristocetin in normal platelets). This normal response to ristocetin but absent response to other agonists is a key diagnostic feature of Glanzmann thrombasthenia.
- Flow Cytometry for GPIIb/IIIa (αIIbβ3) Expression: This technique uses fluorescently labeled antibodies to quantify the amount of αIIbβ3 protein on the platelet surface.
- Type I: Shows a severe reduction (typically <5% of normal) or complete absence of αIIbβ3 expression on the platelet surface.
- Type II: Shows a reduced expression (typically 5-20% of normal) of αIIbβ3. The protein that is present may also be functionally abnormal.
- Variant: May show normal or near-normal levels of αIIbβ3 protein, but functional assays (like LTA) will demonstrate its inability to function properly. Flow cytometry using activation-dependent antibodies might reveal defects in receptor activation.
- Clot Retraction Assay: This test assesses the ability of a blood clot to contract over time, a process that depends on functional platelets and αIIbβ3 binding to fibrin. Clot retraction is typically poor or absent in GT due to the lack of platelet aggregation and their inability to effectively interact with the fibrin mesh.
Genetic Testing
- Gene Sequencing of ITGA2B and ITGB3: Identification of biallelic (homozygous or compound heterozygous) pathogenic mutations in either the ITGA2B or ITGB3 gene. Molecular genetic testing to identify mutations in the genes encoding the αIIb and β3 subunits of the αIIbβ3 integrin can confirm the diagnosis, identify the specific genetic defect, and is useful for carrier testing and prenatal diagnosis in affected families.
Summary of Key Expected Findings
| Test | Expected Finding in GT |
| Platelet Count | Normal |
| Platelet Morphology | Usually Normal |
| PT/aPTT | Normal |
| Bleeding Time | Prolonged |
| PFA-100 | Prolonged or not measurable closure time |
| Platelet Aggregation (LTA) | Absent or severely reduced response to ADP, collagen, epinephrine, thrombin, arachidonic acid |
| Platelet Aggregation (LTA) | Normal or near-normal response to ristocetin |
| Flow Cytometry (GPIIb/IIIa) | Reduced or absent expression (Type I & II), normal levels with functional defect (Variant) |
| Clot Retraction Assay | Poor or absent |
| Genetic Testing | Biallelic pathogenic mutations in ITGA2B or ITGB3 |
Laboratory findings must be interpreted in the context of the patient's clinical history and bleeding symptoms to establish a definitive diagnosis of Glanzmann thrombasthenia and to differentiate it from other platelet function disorders.
Differential Diagnosis for Glanzmann Thormbasthenia
| Disorder | Key Defect | Platelet Count / Morphology | Platelet Aggregation (LTA) | Other Key Features |
| Glanzmann Thrombasthenia (GT) | Defective / deficient αIIbβ3 | Normal | Normal/near-normal to ristocetin; Absent/severely reduced to other agonists | Primarily mucocutaneous bleeding |
| Bernard-Soulier Syndrome (BSS) | Defective / deficient GPIb/IX/V | Low, Large Platelets (Macrothrombocytopenia) | Absent/reduced to ristocetin (not corrected by normal plasma); Normal to other agonists | Macrothrombocytopenia, often more severe bleeding |
| Platelet Storage Pool Deficiencies (SPDs) | Defective granule content or release | Normal | Normal to ristocetin; Impaired secondary wave or reduced to some other agonists | Variable bleeding, may have other associated features (e.g., Hermansky-Pudlak) |
| Wiskott-Aldrich Syndrome (WAS) | Defective WAS protein | Low, Small Platelets (Microthrombocytopenia) | Variable to ristocetin; Variable to other agonists | Eczema, immunodeficiency, microthrombocytopenia |
| Gray Platelet Syndrome (GPS) | Deficiency of alpha granules | Mildly Low, Large, Gray Platelets | Normal to ristocetin; Variable to other agonists | Mild to moderate bleeding, splenomegaly, myelofibrosis (in some) |
| Scott Syndrome | Defective phospholipid scrambling | Normal | Normal | Impaired platelet procoagulant activity |
| Drug-Induced Platelet Dysfunction | Inhibition of platelet function by medications | Normal | Variable to ristocetin (depends on drug); Specific patterns of inhibition to other agonists | History of medication use, symptoms correlate with drug use |
| Uremia | Uremic toxins impairing platelet function | Normal | Normal to ristocetin; Abnormal aggregation (variable patterns) to other agonists | Associated with chronic kidney disease |
| Acquired von Willebrand Syndrome (AVWS) | Decreased/dysfunctional vWF (autoantibodies) | Normal | Reduced to ristocetin (corrects with normal plasma); Normal to other agonists | Later onset, associated with other conditions (lymphoproliferative, autoimmune) |
| Hemophilia A/B | Deficiency of Factor VIII/IX | Normal | Normal | Often presents with deep tissue and joint bleeds |
Treatment and Management
The primary goal of management is to prevent and treat bleeding episodes, as there is currently no cure for this inherited disorder. Treatment strategies are tailored to the severity and frequency of bleeding in each individual.
Management of Acute Bleeding Episodes
The immediate management of active bleeding depends on the site and severity of the bleed.
Local Hemostatic Measures
For mild to moderate mucocutaneous bleeding, local measures are often the first line of treatment.
- Direct Pressure: Applying firm pressure to the bleeding site for an adequate amount of time (e.g., 10-15 minutes for nosebleeds or minor cuts).
- Topical Agents:
- Fibrin Glue: Can be applied to bleeding mucosal surfaces (e.g., gums, nasal mucosa) to promote clot formation.
- Topical Thrombin: May also be used to enhance clotting locally.
- Tranexamic Acid: An antifibrinolytic agent that can be used topically (e.g., as a mouthwash for oral bleeding or nasal spray for epistaxis) to help stabilize clots.
- Nasal Packing: For persistent nosebleeds, nasal packing with absorbable or non-absorbable materials may be necessary.
Platelet Transfusions
This is the primary treatment for significant bleeding episodes or before invasive procedures. Transfused platelets provide a temporary source of functional αIIbβ3, allowing for effective platelet aggregation and hemostasis.
- Considerations:
- Alloimmunization: Repeated platelet transfusions can lead to the development of alloantibodies (antibodies against HLA or platelet-specific antigens) in some patients, resulting in platelet refractoriness (reduced effectiveness of subsequent transfusions).
- HLA-matched platelets: If alloimmunization occurs, HLA-matched or crossmatched platelets may be required to achieve adequate hemostasis, but these can be difficult to obtain.
- Single-donor platelets (apheresis platelets): Using platelets from a single donor can reduce the number of donor exposures and potentially lower the risk of alloimmunization.
Recombinant Activated Factor VIIa (rFVIIa)
This is an alternative hemostatic agent that can be effective in some patients with Glanzmann thrombasthenia, particularly those who have developed alloantibodies and are refractory to platelet transfusions.
rFVIIa works by bypassing the need for platelet aggregation. It binds to tissue factor at the site of injury, leading to the activation of the coagulation cascade and thrombin generation, which can promote hemostasis even in the absence of effective platelet plug formation.
It is often reserved for severe bleeding episodes or before major surgery in patients refractory to platelet transfusions.
Antifibrinolytic Agents
These medications (e.g., tranexamic acid, epsilon-aminocaproic acid) inhibit the breakdown of blood clots (fibrinolysis), helping to maintain hemostasis. It can be given orally or intravenously.
It is often used as adjunctive therapy for mucosal bleeding (e.g., menorrhagia, epistaxis, dental bleeding) and can be helpful in reducing the need for platelet transfusions in some situations.
Prophylactic Measures
Preventing bleeding episodes is a crucial aspect of long-term management.
- Avoidance of Antiplatelet Medications: Patients with Glanzmann thrombasthenia should strictly avoid medications that inhibit platelet function, such as aspirin, nonsteroidal anti-inflammatory drugs (NSAIDs), and other antiplatelet agents (e.g., clopidogrel, ticagrelor).
- Caution with Invasive Procedures: Any invasive medical or dental procedures should be planned carefully, and prophylactic platelet transfusions or other hemostatic agents may be necessary to minimize bleeding risk.
- Menorrhagia Management in Females: Heavy menstrual bleeding can be a significant problem. Management strategies may include:
- Antifibrinolytic agents (e.g., tranexamic acid) during menstruation.
- Hormonal therapies (e.g., oral contraceptives, progestins, levonorgestrel-releasing intrauterine system (LNG-IUS)) to regulate and reduce menstrual flow.
- Iron supplementation to prevent or treat anemia resulting from chronic heavy bleeding.
Management of Alloimmunization and Platelet Refractoriness
Alloimmunization to platelets can complicate management significantly.
- HLA-matched platelets: If antibodies are identified, transfusions with HLA-matched platelets (matched for human leukocyte antigens) may be more effective. Finding HLA-matched donors can be challenging.
- Crossmatched platelets: If HLA-matched platelets are unavailable, crossmatched platelets (tested for compatibility with the patient's serum) may be used.
- Immunosuppressive therapy: In some cases, immunosuppressive medications may be used to reduce the production of alloantibodies.
- Recombinant activated factor VIIa (rFVIIa): As mentioned earlier, rFVIIa can be a valuable alternative in patients with platelet refractoriness.
Supportive Care and Patient Education
- Patient Education: Comprehensive education for patients and their families about Glanzmann thrombasthenia, including recognizing signs of bleeding, avoiding triggers, and understanding treatment options, is crucial for self-management and improving quality of life.
- Psychological and Social Support: Living with a chronic bleeding disorder can have a significant psychological and social impact. Access to support groups and mental health professionals can be beneficial.
- Genetic Counseling: Genetic counseling is important for affected individuals and their families to understand the inheritance pattern, risks for future offspring, and available reproductive options.
- Regular Monitoring: Regular follow-up with a hematologist experienced in bleeding disorders is essential to monitor for bleeding episodes, assess treatment effectiveness, and manage any complications.
Multidisciplinary Approach
Optimal management of Glanzmann thrombasthenia often requires a multidisciplinary team, including hematologists, nurses specializing in bleeding disorders, dentists, gynecologists, orthopedic surgeons (if joint bleeds occur), genetic counselors, and social workers.
Frequently Asked Questions (FAQs)
What do Bernard-Soulier syndrome and Glanzmann's thrombasthenia have in common?
Bernard-Soulier syndrome (BSS) and Glanzmann's thrombasthenia (GT) are both rare, inherited bleeding disorders resulting from defects in platelet surface glycoproteins, leading to impaired platelet function and a lifelong history of mucocutaneous bleeding. In both conditions, specialized laboratory tests, particularly platelet aggregation studies and flow cytometry, are crucial for diagnosis, revealing abnormalities in how platelets interact and the presence or absence of specific glycoproteins. While GT is characterized by a defect in αIIbβ3 affecting platelet aggregation to most agonists but not ristocetin, BSS involves a deficiency in the GPIb/IX/V complex, impairing platelet adhesion and causing a reduced or absent response to ristocetin.
What age is Glanzmann thrombasthenia diagnosed?
Glanzmann thrombasthenia (GT) is often diagnosed early in life, typically in infancy or early childhood. This is because the symptoms, such as easy bruising, frequent nosebleeds, bleeding gums, and prolonged bleeding after minor injuries or procedures like circumcision, usually manifest from birth or shortly thereafter.
According to research, over 80% of individuals with GT are diagnosed before the age of 14, and many are diagnosed before the age of 5. However, in milder cases, the diagnosis might be delayed until adulthood, sometimes triggered by a significant bleeding event like trauma or surgery.
What is the life expectancy of Glanzmann thrombasthenia?
With proper management and supportive care, most individuals with Glanzmann thrombasthenia (GT) can expect to live a normal life expectancy. While GT is a lifelong condition with no cure, advancements in treatment, particularly the availability of platelet transfusions and recombinant activated factor VIIa (rFVIIa), have significantly improved the prognosis. Although severe bleeding episodes can be life-threatening, especially during major events like surgery or childbirth, proactive management and avoidance of bleeding triggers help patients lead full and active lives. In some individuals, the frequency and severity of bleeding episodes may even decrease with age.
Disclaimer: This article is intended for informational purposes only and is specifically targeted towards medical students. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Goldberg S, Hoffman J. Clinical Hematology Made Ridiculously Simple, 1st Edition: An Incredibly Easy Way to Learn for Medical, Nursing, PA Students, and General Practitioners (MedMaster Medical Books). 2021.
- Krause KA, Graham BC. Glanzmann Thrombasthenia. [Updated 2023 Aug 28]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK538270/
- Botero, J. P., Lee, K., Branchford, B. R., Bray, P. F., Freson, K., Lambert, M. P., Luo, M., Mohan, S., Ross, J. E., Bergmeier, W., Di Paola, J., & ClinGen Platelet Disorder Variant Curation Expert Panel (2020). Glanzmann thrombasthenia: genetic basis and clinical correlates. Haematologica, 105(4), 888–894. https://doi.org/10.3324/haematol.2018.214239
- Poon, M. C., Di Minno, G., d'Oiron, R., & Zotz, R. (2016). New Insights Into the Treatment of Glanzmann Thrombasthenia. Transfusion medicine reviews, 30(2), 92–99. https://doi.org/10.1016/j.tmrv.2016.01.001
- Mathews N, Rivard GE, Bonnefoy A. Glanzmann Thrombasthenia: Perspectives from Clinical Practice on Accurate Diagnosis and Optimal Treatment Strategies. J Blood Med. 2021;12:449-463 https://doi.org/10.2147/JBM.S271744