Procedure At-A-Glance
The G6PD test screens for glucose-6-phosphate dehydrogenase deficiency, the most common inherited enzyme disorder, affecting around 400 million people worldwide [3]. The fluorescent spot test (FST) is a fast, low-cost qualitative G6PD test that uses ultraviolet light to detect NADPH, the molecule produced by a working G6PD enzyme [8].
- Reconstitute the G6PD reagent. Add 5 mL of dilution buffer to the freeze-dried reagent and let it dissolve for 30 minutes.
- Reconstitute the quality control vials. Add 50 µL of distilled water to each freeze-dried QC powder (normal, intermediate, deficiency) and let dissolve for 30 minutes.
- Label four microcentrifuge tubes: Normal Control, Intermediate Control, Deficiency Control, and Patient.
- Add 100 µL of G6PD reagent to each tube.
- Add 5 µL of the matching QC material or patient sample to each tube and mix thoroughly.
- Incubate 10 minutes at room temperature.
- Spot 10 µL of each mixture onto labeled filter paper.
- Air-dry at room temperature for about 1 hour.
- View the dried spots under a long-wave UV lamp in a darkened room and compare patient fluorescence with the QC spots.
Why This Test Matters
Imagine a patient with malaria who is about to receive a single-dose tablet to wipe out dormant liver parasites. If that patient has G6PD deficiency, the same tablet can rupture their red blood cells within hours. The G6PD test exists to prevent exactly this kind of preventable harm. It also guides decisions in newborns at risk of severe jaundice, in patients with unexplained hemolytic anemia, and in family members of affected individuals.
This article walks through the principle, protocol, and interpretation of the fluorescent spot test, then sets it in the wider context of how G6PD deficiency is diagnosed today.
What is G6PD deficiency?
Glucose-6-phosphate dehydrogenase, or G6PD, is the gatekeeper enzyme of the pentose phosphate pathway in red blood cells. It produces NADPH, which keeps glutathione in its reduced (active) form. Reduced glutathione is the red cell's main shield against oxidative damage [3].
When G6PD activity is low, this defense fails. Red cells exposed to oxidants like certain drugs, infections, or fava beans, the cells accumulate damage, lose membrane integrity, and break apart in a process called hemolysis [6]. Mature red cells are uniquely vulnerable because they have no mitochondria and depend entirely on this single pathway for NADPH.
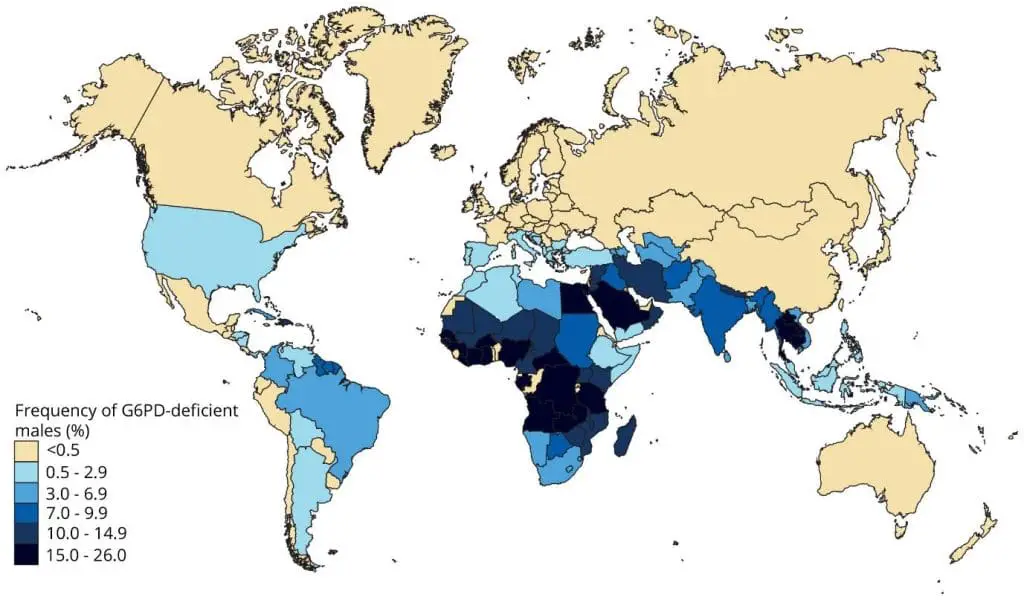
G6PD deficiency affects roughly 400 million people worldwide, with the highest prevalence in sub-Saharan Africa, the Mediterranean, the Middle East, and Southeast Asia which broadly mirroring historical malaria endemicity [3]. The geographic overlap is not a coincidence; G6PD deficiency offers some protection against severe malaria, which is why the trait persists.
How It Is Inherited
G6PD deficiency is X-linked recessive. The G6PD gene sits on the X chromosome, so inheritance follows a sex-linked pattern.
- Males (XY) are hemizygous. A single defective gene produces full deficiency.
- Females (XX) can be homozygous (two defective copies, fully deficient), heterozygous carriers (one defective, one normal), or unaffected (two normal copies).

Heterozygous females deserve special attention. Because of X-inactivation, one X chromosome is randomly silenced in each cell during early development. The result is a mosaic: some red cells have normal G6PD activity, others have deficient activity. The mix varies from woman to woman, which is why heterozygotes can range from clinically silent to as symptomatic as a deficient male [3,7].
Clinical Phenotypes
G6PD deficiency presents in four main ways:
- Acute hemolytic anemia triggered by an oxidant drug, infection, or fava bean ingestion.
- Neonatal jaundice, which can be severe enough to cause kernicterus if not treated promptly.
- Favism, a specific form of acute hemolysis after eating fava beans.
- Chronic non-spherocytic hemolytic anemia (CNSHA), a rare lifelong form caused by Class A variants under the 2024 WHO classification [1].
Symptoms during a hemolytic crisis include pale skin, jaundice, fatigue, dark urine (hemoglobinuria), shortness of breath, abdominal or back pain, and fever.
The 2024 WHO Variant Classification
The WHO updated the G6PD variant classification system (endorsed by the WHO Malaria Policy Advisory Group in March 2022, and formally published and integrated into guidelines in 2024) [1]. The new system has four classes:
- Class A — variants that cause chronic non-spherocytic hemolytic anemia.
- Class B — common polymorphic variants (the previous classes II and III, now merged) that cause severe neonatal jaundice and acute hemolysis triggered by drugs, infection, or fava beans.
- Class C — variants with normal or near-normal activity, no clinical disease.
- Class U — variants of uncertain clinical significance.
The merger of the old classes II and III emphasizes that any common variant can produce severe hemolysis under the right trigger [1,7].
G6PD Deficiency
| Class | Enzyme Activity | Clinical Presentation | |
|---|---|---|---|
| A Severe | < 20% of normal | Chronic non-spherocytic hemolytic anemia (CNSHA) — hemolysis occurs constitutively, even in the absence of an exogenous trigger. | |
| B Moderate | < 45% of normal | Acute hemolysis triggered by oxidative stressors — including drugs, infections, or fava bean ingestion. Asymptomatic at baseline. | |
| C Normal | 60 – 150% of normal | No clinically significant hemolysis. Enzyme activity within or above the reference range. | |
| U Uncertain | Any level | Variant of uncertain clinical significance — insufficient evidence to classify clinical risk based on current data. |
Treatment and Management
There is no cure. Management rests on avoiding triggers and treating crises when they occur. Key trigger categories are:
- Oxidant drugs: primaquine, tafenoquine, dapsone, rasburicase, sulfonamides, nitrofurantoin, methylene blue, and high-dose aspirin.
- Foods: fava beans (the strongest food trigger).
- Infections: any febrile illness can precipitate hemolysis.
Patients with severe acute hemolysis may need fluid support, monitoring for renal injury from hemoglobinuria, and occasionally blood transfusion. Newborns with severe jaundice may require phototherapy or exchange transfusion to prevent kernicterus [6].
Principle of G6PD Fluorescent Spot Test
The G6PD test exploits a simple chemistry: when G6PD is working, it produces NADPH, and NADPH glows green under long-wave UV light [8].
The reaction is:
G6P + NADP⁺ → 6-phosphogluconate + NADPH + H⁺
A blood sample is mixed with G6P and NADP⁺. After incubation, a drop is spotted onto filter paper and dried. Under UV light, normal samples fluoresce bright green. Deficient samples fluoresce faintly or not at all because little NADPH was made [8]. The amount of NADPH is directly proportional to G6PD activity, which makes the test a useful screening tool though it is qualitative, not numerical.
Methods vary slightly by manufacturer, so always follow the kit insert.
When the G6PD Test Is Ordered
Modern indications for the G6PD test include:
- Before prescribing primaquine or tafenoquine for P. vivax or P. ovale malaria. WHO's 2024 malaria guidelines specifically call for near-patient G6PD testing to guide anti-relapse treatment, with quantitative testing strongly preferred before single-dose tafenoquine [2].
- Newborn screening in countries where G6PD deficiency is common (Malaysia, Singapore, Greece, parts of the United States and the Middle East).
- Investigation of unexplained hemolytic anemia or jaundice, particularly after a known trigger.
- Family screening when a relative is affected.
- Before drugs known to trigger hemolysis in patients with relevant ancestry or symptoms.
Materials
- 5 µL of whole blood anticoagulated with EDTA (purple-top tube), heparin (green-top), or acid-citrate-dextrose (ACD, yellow-top) [9]; or, for newborn screening, cord blood spotted onto filter paper.
- Dilution buffer
- G6PD reagent (freeze-dried substrate containing G6P and NADP⁺)
- Distilled water
- QC vials: normal, intermediate, and deficiency controls
- Filter paper suitable for dried blood spots (e.g., Whatman Grade 903 or equivalent neonatal screening paper)
- Microcentrifuge tubes
- Long-wave UV lamp / UV viewing box
Protocol of G6PD Test
- Prepare the G6PD reagent stock. Add 5 mL of dilution buffer to the freeze-dried reagent vial. Let it dissolve fully for 30 minutes. The reconstituted reagent is stable for 4 weeks at +4 °C and 2 months at −20 °C.
- Prepare the QC solutions. Add 50 µL of distilled water to each freeze-dried QC vial (normal, intermediate, deficiency). Let dissolve for 30 minutes.
- Label 4 microcentrifuge tubes: Normal Control, Intermediate Control, Deficiency Control, Patient.
- Add 100 µL of G6PD reagent to each labeled tube.
- Add 5 µL of the matching QC or patient sample to each tube. Mix thoroughly.
- Incubate at room temperature for 10 minutes.
- Spot 10 µL from each tube onto the matching labeled section of filter paper.
- Air-dry at room temperature for about 1 hour.
- View under a long-wave UV lamp in a darkened room.
- Compare the patient spot with the three QC spots and report.
Interpretation of G6PD Test
The fluorescent spot test gives a visual, qualitative result. The patient's spot is compared with the three QC spots run in the same batch.
- Normal G6PD activity — bright green fluorescence. NADPH is being produced normally [8].
- Intermediate G6PD activity — dim or yellowish-green fluorescence. NADPH production is reduced. Common in heterozygous female carriers and some milder variants.
- Deficient G6PD activity — no fluorescence or barely visible fluorescence. NADPH production has effectively stopped.
Because the test relies on visual judgment, two trained observers should ideally agree on the result before reporting.
Troubleshooting
The fluorescent spot test is reliable when the conditions are right, but several factors can throw it off [3,6]:
- Sample handling: delayed processing, hemolyzed samples, or incorrect anticoagulant can all degrade enzyme activity.
- Reagent quality: expired or improperly stored reagent reduces sensitivity.
- Incubation time: too short underestimates activity; too long can give a misleading positive.
- UV source: low-intensity or wrong-wavelength UV makes intermediate results harder to read.
- Recent hemolytic crisis: young red cells made after the crisis carry near-normal enzyme activity. The patient's older deficient cells are gone, so the test can read falsely normal. Wait several weeks before retesting.
- Recent blood transfusion: donor red cells with normal G6PD activity mask the patient's enzyme status. Wait at least 30 days before testing [6].
- Heterozygous females: the most clinically important limitation. The mosaic of normal and deficient cells in a carrier produces enough NADPH to mask the deficient population, and the FST often returns a "normal" result even when a clinically significant fraction of cells is deficient [3,7]. This matters most before prescribing tafenoquine or primaquine.
How the G6PD Test Compares with Other Methods
The fluorescent spot test is one of four laboratory approaches. Each has trade-offs, and modern practice often combines them.
| Method | Principle | Strengths | Weaknesses |
|---|---|---|---|
| Fluorescent Spot Test (FST) Qualitative | Visual detection of NADPH fluorescence under UV light. [8] | Cheap, fast, suited to mass screening and low-resource settings; good sensitivity for severe deficiency. | Misses many heterozygous females; false-negatives after hemolysis or transfusion; subjective reading. |
| Quantitative Spectrophotometric Assay Quantitative · IU/gHb Gold Standard | Measures rate of NADPH formation by absorbance, reported in IU/gHb. [4] | Precise; detects mild and intermediate deficiency; identifies female carriers more reliably. | Requires lab equipment and trained staff; hours-to-days turnaround; affected by recent hemolysis or transfusion. |
| Point-of-Care Quantitative Biosensor Quantitative · Bedside | Handheld biosensor reports enzyme activity (often with hemoglobin) within minutes. WHO prequalified the first such device in December 2024. [5] | Numerical result at the bedside; supports same-visit primaquine or tafenoquine decisions. | More expensive than FST; performance varies by device; can miss some carriers; temperature-sensitive. |
| Molecular Genetic Testing Genotype | Sequences the G6PD gene to identify specific variants. [1] | Definitive genotype; unaffected by recent hemolysis or transfusion; best for confirming carriers and classifying variants. | Expensive; slow (days to weeks); identifies the variant but does not directly measure enzyme activity. |
FST vs. Quantitative Testing
While the FST remains a workhorse for public health and neonatal screening, in the context of P. vivax malaria management, point-of-care quantitative biosensors have largely displaced the FST. Following the strong recommendation for quantitative testing prior to single-dose tafenoquine, these biosensors are now the preferred standard in malaria clinics [10].
Frequently Asked Questions (FAQs)
What is the G6PD blood spot test and how does it work?
The G6PD fluorescent spot test (FST) is a simple, rapid screen for G6PD deficiency. It works by mixing blood with G6P and NADP⁺. Functional G6PD converts NADP⁺ to NADPH, which fluoresces green under UV light. Bright green fluorescence indicates normal activity; faint or absent fluorescence suggests deficiency.
What is the normal fluorescence look like in a G6PD test?
Normal fluorescence is a bright green glow under long-wave UV light. This green color comes from NADPH, the product of the G6PD-catalyzed reaction. Patient spots are always compared against normal, intermediate, and deficient quality control spots run in the same batch.
Can the G6PD test give a wrong result?
Yes. False-negatives are most common after a recent hemolytic crisis (because surviving young red cells have near-normal activity), after blood transfusion (donor cells mask deficient ones for about 30 days), and in heterozygous female carriers whose mosaic of normal and deficient cells fools the qualitative readout. False-positives can occur from sample handling errors or borderline mild deficiency. When the result will guide a decision like tafenoquine prescribing, a quantitative test is preferred.
Why does the test miss many female carriers?
Females have two X chromosomes, but only one is active in each cell because of X-inactivation. A heterozygous carrier has a mosaic of red cells: some with normal G6PD, some with deficient G6PD. The normal cells produce enough NADPH to make the spot fluoresce green, hiding the deficient population. Quantitative or genetic testing is more reliable for carrier detection.
What conditions can mimic G6PD deficiency?
When a patient presents with hemolysis or jaundice, the differential includes pyruvate kinase deficiency, hereditary spherocytosis, sickle cell disease, thalassemia, autoimmune hemolytic anemia, hemolytic disease of the newborn, Gilbert syndrome, and other causes of hemolytic anemia. Clinical history, peripheral blood smear, and targeted enzyme or molecular testing usually clarify the cause.
What can trigger a hemolytic crisis in G6PD deficiency?
The three major trigger categories are oxidant drugs (primaquine, tafenoquine, dapsone, rasburicase, sulfonamides, nitrofurantoin, methylene blue), infections (which generate reactive oxygen species during the immune response), and fava beans (favism). Aspirin and high-dose vitamin C can also be a problem in some variants. Patients should always carry a list of drugs to avoid.
Why is G6PD testing required before tafenoquine and primaquine?
Both drugs eliminate dormant P. vivax and P. ovale parasites in the liver, but both are powerful oxidants. Giving them to a G6PD-deficient patient can trigger severe, sometimes life-threatening hemolysis. Tafenoquine is the larger concern because it is given as a single long-acting dose that cannot be stopped once swallowed. WHO's 2024 malaria guidelines recommend confirming adequate G6PD activity, ideally with a quantitative or semi-quantitative test, before either drug [2].
Glossary of Related Medical Terms
- G6PD (glucose-6-phosphate dehydrogenase): An enzyme inside red blood cells that helps the cell make NADPH, the molecule that protects the cell from oxidative damage.
- NADPH (nicotinamide adenine dinucleotide phosphate hydrogen): The "shielding" molecule that recycles glutathione, the red cell's main antioxidant defense.
- Pentose phosphate pathway: The metabolic pathway, started by G6PD, that produces NADPH in red blood cells.
- Hemolysis: Premature breakdown of red blood cells, releasing hemoglobin into the bloodstream.
- Hemolytic crisis: A sudden episode of large-scale red cell destruction, often triggered by drugs, infection, or fava beans in G6PD deficiency.
- Oxidative stress: Damage caused by reactive oxygen species when antioxidant defenses are overwhelmed.
- Hemizygous: Having only one copy of a gene, as males do for X-linked genes.
- Heterozygous carrier (female): A female with one normal and one defective G6PD gene, who has a mixed population of normal and deficient red cells due to X-inactivation.
- X-inactivation (lyonization): The random switching off of one X chromosome in each female cell, producing a mosaic pattern.
- Qualitative test: A test that gives a yes/no or visual category result rather than a precise number.
- Quantitative assay: A test that gives an exact numerical value, in this case enzyme activity in international units per gram of hemoglobin (IU/gHb).
- Fluorescence: Light emitted by a substance after absorbing UV or other light.
- Filter paper / Guthrie card: Absorbent paper used to spot dried blood for screening tests.
- Hypnozoite: The dormant liver-stage form of P. vivax and P. ovale malaria parasites that causes relapses.
- Tafenoquine: A single-dose antimalarial used to clear hypnozoites; requires G6PD testing before use.
- Primaquine: A 14-day antimalarial used to clear hypnozoites; can cause severe hemolysis in G6PD-deficient patients.
Disclaimer: This protocol is for educational purposes only. Local laboratory standard operating procedures take precedence. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. Always consult a qualified healthcare professional for clinical decision-making. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Luzzatto, L., Bancone, G., Dugué, P. A., Jiang, W., Minucci, A., Nannelli, C., Pfeffer, D., Prchal, J., Sirdah, M., Sodeinde, O., Vulliamy, T., Wanachiwanawin, W., Cunningham, J., & Bosman, A. (2024). New WHO classification of genetic variants causing G6PD deficiency. Bulletin of the World Health Organization, 102(8), 615–617. https://doi.org/10.2471/BLT.23.291224
- World Health Organization. (2025). WHO guidelines for malaria, 13 August 2025. World Health Organization.
- Luzzatto, L., Ally, M., & Notaro, R. (2020). Glucose-6-phosphate dehydrogenase deficiency. Blood, 136(11), 1225–1240. https://doi.org/10.1182/blood.2019000944
- Sharma, U., Mishra, S., Gautam, N., & Gupta, B. K. (2020). Qualitative and quantitative assay of glucose 6 phosphate dehydrogenase in patients attending tertiary care center. BMC research notes, 13(1), 298. https://doi.org/10.1186/s13104-020-05145-8
- Zailani, M. A. H., Raja Sabudin, R. Z. A., Abdul Jalil, D., Ithnin, A., Ayub, N. A., Alauddin, H., Jalil, N., Md Fauzi, A., Cheah, F. C., Lim, L. S., Safian, N., Yusuf, M. M., & Othman, A. (2023). Evaluation of quantitative point-of-care test for measurement of glucose-6-phosphate dehydrogenase enzyme activity in Malaysia. The Malaysian journal of pathology, 45(1), 31–41.
- Mak GK, Shah M. Glucose-6-Phosphate Dehydrogenase Deficiency. [Updated 2025 Nov 30]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2026 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK470315/
- Nannelli, C., Bosman, A., Cunningham, J., Dugué, P. A., & Luzzatto, L. (2023). Genetic variants causing G6PD deficiency: Clinical and biochemical data support new WHO classification. British journal of haematology, 202(5), 1024–1032. https://doi.org/10.1111/bjh.18943
- Beutler E. (1966). A series of new screening procedures for pyruvate kinase deficiency, glucose-6-phosphate dehydrogenase deficiency, and glutathione reductase deficiency. Blood, 28(4), 553–562. https://doi.org/10.1182/blood.V28.4.553.553
- Roper, D., Layton, M., Rees, D., Lambert, C., Vulliamy, T., De la Salle, B., D'Souza, C., & British Society for Haematology (2020). Laboratory diagnosis of G6PD deficiency. A British Society for Haematology Guideline. British journal of haematology, 189(1), 24–38. https://doi.org/10.1111/bjh.16366
- Zailani, M. A. H., Raja Sabudin, R. Z. A., Ithnin, A., Alauddin, H., Sulaiman, S. A., Ismail, E., & Othman, A. (2023). Population screening for glucose-6-phosphate dehydrogenase deficiency using quantitative point-of-care tests: a systematic review. Frontiers in genetics, 14, 1098828. https://doi.org/10.3389/fgene.2023.1098828