TL;DR
Diamond-Blackfan Anemia or DBA is a rare, congenital blood disorder where the bone marrow fails to produce red blood cells. It is also known as congenital hypoplastic anemia.
- Causes ▾: The core cause is a defect in ribosome biogenesis, primarily due to mutations in ribosomal protein genes like RPS19. It is most often inherited in an autosomal dominant pattern or occurs sporadically.
- Clinical Presentation ▾: Symptoms, including severe pallor and fatigue, appear within the first year of life. Approximately 50% of patients also have associated physical malformations, such as abnormalities of the thumb, head, or heart.
- Diagnosis ▾: Diagnosis is made through a combination of severe macrocytic anemia, very low reticulocyte count, and a bone marrow biopsy showing a selective absence of red blood cell precursors. Supportive lab findings include elevated eADA and HbF levels. Genetic testing can confirm the diagnosis.
- Treatment ▾: First-line treatment is corticosteroids. For patients who don’t respond, red blood cell transfusions are used, which require iron chelation therapy to prevent iron overload. The only known cure is a hematopoietic stem cell transplant (HSCT).
- Prognosis ▾: The prognosis has improved with modern treatment, but it remains a chronic condition with a significant long-term risk of developing complications like iron overload and an increased risk of cancers such as Myelodysplastic Syndrome (MDS) and Acute Myeloid Leukemia (AML).
*Click ▾ for more information
Introduction
Diamond-Blackfan Anemia (DBA) is a rare inherited bone marrow failure syndrome characterized by a profound deficit in erythropoiesis, leading to severe macrocytic anemia typically presenting in infancy or early childhood.
As a prototypical ribosomopathy, Diamond-Blackfan anemia (DBA) is fundamentally a disorder of ribosome biogenesis, affecting the production and function of hematopoietic stem and progenitor cells, particularly the erythroid lineage.
Understanding the intricate genetic and cellular mechanisms underlying Diamond-Blackfan anemia (DBA) is crucial for medical students and professionals to appreciate its complex clinical presentation, diagnostic challenges, and evolving therapeutic strategies.
Causes and Genetics of Diamond-Blackfan Anemia (DBA)
The fundamental cause of Diamond-Blackfan Anemia is a defect in ribosome biogenesis, which is the process of creating ribosomes – the cellular machinery responsible for protein synthesis. This is a crucial function for all cells, but for reasons not yet fully understood, erythroid progenitor cells (the precursors to red blood cells) are particularly sensitive to this defect.
Approximately 60-70% of Diamond-Blackfan anemia (DBA) cases are associated with heterozygous mutations in genes encoding ribosomal proteins (RPs), with RPS19 being the most commonly affected gene. Other frequently mutated genes include RPL5, RPL11, RPS7, and RPS10. These mutations result in haploinsufficiency, where a single functional copy of the gene is insufficient to produce adequate amounts of the ribosomal protein, which in turn disrupts the assembly of the small (40S) or large (60S) ribosomal subunits.
Regarding inheritance, the majority of familial DBA cases follow an autosomal dominant inheritance pattern. This means that a person needs to inherit only one copy of the mutated gene from a parent to be affected. However, it is important to note that many cases, roughly 40-50%, are sporadic, meaning they result from a new, random mutation in the affected individual and are not inherited from a parent.
Pathophysiology: A Ribosomopathy in Focus
The hallmark of Diamond-Blackfan anemia (DBA) pathophysiology is a defect in the synthesis of ribosomes, the cellular machinery responsible for protein production.
The disrupted ribosome biogenesis leads to a cascade of cellular stress responses. The accumulation of unassembled ribosomal components, particularly the 5S and 28S rRNAs, activates the ribosomal stress pathway. This pathway involves the sequestration of a protein called MDM2 by free ribosomal proteins (e.g., RPL5 and RPL11).
MDM2 is a ubiquitin ligase that normally targets the tumor suppressor protein p53 for degradation. By sequestering MDM2, the defective ribosome biogenesis leads to increased stabilization and activation of p53.
The central role of p53 activation in Diamond-Blackfan anemia (DBA) is profound. Activated p53 triggers cell cycle arrest and apoptosis (programmed cell death), particularly in highly proliferative cells like erythroid progenitors in the bone marrow.
This selective pressure on erythroid cells explains the characteristic pure red cell aplasia observed in Diamond-Blackfan anemia (DBA), while other hematopoietic lineages, such as myeloid and megakaryocytic cells, are often less affected. The p53-mediated apoptosis of erythroid progenitors results in the severe anemia that defines the disease.
While p53 activation is a dominant model, other mechanisms likely contribute to Diamond-Blackfan anemia (DBA) pathophysiology. These include impaired translation of specific mRNAs and dysregulation of developmental pathways.
Recent research has also highlighted the role of micro-RNAs and epigenetic modifications in modulating the severity and clinical phenotype of Diamond-Blackfan anemia (DBA), suggesting a more complex regulatory network beyond simple p53-mediated apoptosis.
Clinical Manifestations and Symptoms of Diamond-Blackfan Anemia (DBA)
The signs and symptoms of Diamond-Blackfan Anemia typically appear very early in life, with the onset of the disease usually occurring within the first year. Most patients are diagnosed by 6 months of age, and over 90% by the time they are 1 year old. The initial presentation is often subtle but progresses to noticeable signs of severe anemia.
The hematological symptoms are a direct result of the bone marrow’s inability to produce an adequate number of red blood cells. These symptoms include:
- Pallor: The most common sign, reflecting the lack of red blood cells. This may be observed as a pale complexion or paleness of the nail beds and inner eyelids.
- Lethargy and Fatigue: Infants with severe anemia will often seem tired, lack energy, and be less active than their peers.
- Irritability: An anemic infant may be fussy and difficult to comfort.
- Poor Feeding and Failure to Thrive: The lack of oxygen-carrying capacity can lead to difficulty feeding and failure to gain weight at a normal rate.
- Tachycardia: In an attempt to compensate for the low oxygen levels, the heart rate increases, which can be noticed by a faster pulse.
In addition to these symptoms of anemia, a significant feature of Diamond-Blackfan anemia (DBA) is the presence of physical and congenital anomalies, which are seen in approximately 50% of patients.
Ribosomes are essential for protein synthesis in all cells, and the stress caused by their defective production during embryonic development disproportionately impacts rapidly dividing and differentiating cells, leading to a wide array of congenital anomalies.
These malformations are a key clinical feature, as they are present in approximately 50% of patients. They can affect multiple body systems and often include:
- Craniofacial Anomalies: A significant number of patients may have distinct facial features, such as a broad, flat nasal bridge, hypertelorism (widely spaced eyes), microcephaly (a smaller-than-normal head), or a cleft palate.
- Upper Limb Abnormalities: This is one of the most classic and frequently observed malformations in Diamond-Blackfan anemia (DBA). These can range from a triphalangeal thumb (a thumb with three bones instead of the usual two) to complete absence or hypoplasia (underdevelopment) of the thumb. Other radial ray abnormalities, affecting the forearm bones, may also be present.
- Cardiac Defects: Heart abnormalities are also common and can include atrial septal defects (ASD), ventricular septal defects (VSD), and tetralogy of Fallot.
- Urogenital Malformations: Abnormalities of the kidneys or urinary tract can occur in some patients.
These associated malformations are an important clue for diagnosis, especially in infants who may not yet show the full hematological picture. The presence of these physical signs, along with severe anemia, strongly suggests a diagnosis of Diamond-Blackfan anemia (DBA).
While the anemia is the primary hematologic concern, patients with Diamond-Blackfan anemia (DBA) also have a significantly increased risk of developing other hematologic malignancies, most notably myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML), as well as solid tumors. This predisposition to malignancy further underscores the role of ribosomal and cellular stress in compromising genomic stability.
Laboratory Investigations and Diagnosis of Diamond-Blackfan anemia (DBA)
The diagnosis of Diamond-Blackfan anemia (DBA) is based on a combination of clinical, hematologic, and genetic findings.
Clinical suspicion
The presence of severe macrocytic anemia in an infant with a reticulocyte count that is disproportionately low for the degree of anemia should raise suspicion. The presence of congenital anomalies, a family history of Diamond-Blackfan anemia (DBA), or a history of recurrent transfusions further supports the diagnosis.
Hematologic evaluation
Complete Blood Count (CBC)
In a patient with Diamond-Blackfan anemia (DBA), this will reveal a severe macrocytic anemia. This means that the red blood cells are significantly larger than normal. A key finding is a low or normal reticulocyte count (reticulocytopenia), which indicates that the bone marrow is not producing new red blood cells to compensate for the anemia. In contrast to other bone marrow failure syndromes, the white blood cell and platelet counts are usually within the normal range, a hallmark of a “pure red cell aplasia.”
Bone Marrow Aspiration/Biopsy
The classic finding is a pure red cell aplasia, meaning a severe lack of erythroid precursor cells in an otherwise normal cellular marrow.
Other Laboratory Findings
While not definitive on their own, two specific laboratory markers can provide strong supportive evidence for a Diamond-Blackfan anemia (DBA) diagnosis.
- Elevated erythrocyte adenosine deaminase (eADA) levels: This enzyme is often elevated in the red blood cells of DBA patients for reasons that are not fully understood, but its presence is highly suggestive of the condition.
- Elevated fetal hemoglobin (HbF) levels: Many patients with DBA have abnormally high levels of HbF, which is a type of hemoglobin typically found in the fetus.
Genetic testing
Molecular genetic testing for mutations in ribosomal protein genes is the gold standard for confirming the diagnosis. This involves targeted gene panels or whole exome sequencing. Identification of a pathogenic variant in a known DBA-associated gene confirms the diagnosis. As mentioned previously, mutations in the RPS19 gene are the most common cause, but testing panels now include a wide range of genes known to be associated with the disease.
Differential Diagnosis of Diamond-Blackfan Anemia (DBA)
The key to a correct diagnosis of Diamond-Blackfan anemia (DBA) lies in a combination of the clinical picture, laboratory findings, and ruling out other bone marrow failure syndromes. The main conditions that need to be considered are:
Aplastic Anemia
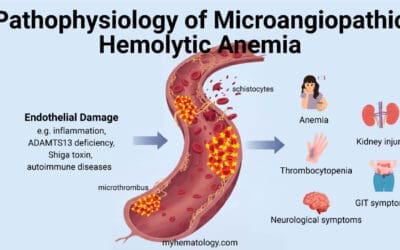
This is perhaps the most important differential diagnosis to consider. While both Diamond-Blackfan anemia (DBA) and aplastic anemia involve bone marrow failure, the key difference is the cell lines affected.
Diamond-Blackfan anemia (DBA) is a pure red cell aplasia, meaning only the red blood cell precursors are significantly reduced, while white blood cells and platelets are typically normal.
In contrast, aplastic anemia is characterized by pancytopenia, a deficiency of all three major blood cell types (red blood cells, white blood cells, and platelets). A bone marrow biopsy is the definitive tool for distinguishing between these two conditions.
Transient Erythroblastopenia of Childhood (TEC)
As the name suggests, this is a temporary condition that also results in a pure red cell aplasia. However, it is an acquired, not congenital, disease.
The primary distinguishing features are its age of onset (typically between 6 months and 4 years), the absence of congenital physical anomalies, and the fact that it spontaneously resolves on its own, usually within a few weeks or months, without specific treatment.
Fanconi Anemia
This is another inherited bone marrow failure syndrome. Like Diamond-Blackfan anemia (DBA), it is associated with various congenital malformations, including skeletal, renal, and gastrointestinal abnormalities.
However, Fanconi Anemia typically progresses to pancytopenia over time, a feature that separates it from the pure red cell aplasia of Diamond-Blackfan anemia (DBA). The definitive diagnostic test for Fanconi Anemia is a chromosomal fragility study, which looks for chromosome breakage when exposed to certain chemicals.
Other Pure Red Cell Aplasias
There are other, rarer causes of pure red cell aplasia that need to be considered. For example, a temporary aplasia can be caused by an infection with Parvovirus B19, which targets and destroys red blood cell precursors. This can be ruled out with serology testing to look for signs of a recent infection.
This table summarizes the key features that distinguish Diamond-Blackfan Anemia from other conditions that may present with similar symptoms.
| Condition | Key Distinguishing Features | Affected Cell Lines | Associated Physical Anomalies | Definitive Diagnostic Test |
| Diamond-Blackfan Anemia (DBA) | Congenital onset, usually within the first year of life. Pure red cell aplasia. | Red blood cells only. | Present in ~50% of patients (e.g., thumb abnormalities, craniofacial defects). | Genetic testing for ribosomal protein mutations. |
| Aplastic Anemia | Acquired or inherited condition. Gradual onset. | All three major cell lines are affected (pancytopenia). | Typically none for acquired form; inherited forms (e.g., Fanconi Anemia) have malformations. | Bone marrow biopsy showing hypocellularity and absence of all three cell lines. |
| Transient Erythroblastopenia of Childhood (TEC) | Acquired condition, typically resolves spontaneously within weeks or months. | Red blood cells only. | None. | Spontaneous resolution; absence of genetic mutations. |
| Fanconi Anemia | Inherited, often progressive. Onset can be later in childhood. | Initially pure red cell aplasia, but progresses to pancytopenia. | Present in most cases (e.g., thumb defects, short stature, café-au-lait spots). | Chromosomal fragility testing. |
| Parvovirus B19 Infection | Acute onset, often following a viral illness. Self-limiting. | Red blood cells only (transient). | None. | Viral serology or PCR for Parvovirus B19. |
Treatment and Management of Diamond-Blackfan Anemia (DBA)
The treatment and management of Diamond-Blackfan Anemia (DBA) are focused on correcting the severe anemia, manage iron overload from chronic transfusions, and monitor for disease-related complications and malignancies. The approach is a step-wise process, starting with the least invasive treatments and moving to more definitive, though riskier, options.
First-line Treatment: Corticosteroid Therapy
This is the first and most common line of treatment, often started as soon as a definitive diagnosis is made.
Corticosteroids, such as prednisone, induce a remission in a significant number of patients, approximately 80%. The exact mechanism by which corticosteroids stimulate red blood cell production in Diamond-Blackfan Anemia (DBA) is not fully understood, but it is thought to reduce apoptosis (programmed cell death) in the erythroid precursors.
If a patient responds to corticosteroids, the dosage is carefully tapered to the lowest possible dose that maintains a stable hemoglobin level to minimize long-term side effects.
Second-line Treatment: Red Blood Cell Transfusions
For patients who do not respond to corticosteroids, or for those who become “steroid-dependent” (meaning they require a high dose to remain stable), regular red blood cell transfusions become necessary.
Transfusions provide the body with the red blood cells it cannot produce on its own. While transfusions are effective at managing the anemia, lifelong transfusion therapy presents a major long-term complication: iron overload. The body has no efficient way to excrete the excess iron from the transfused blood, which accumulates in organs like the heart, liver, and endocrine glands, leading to significant damage.
Iron Chelation Therapy
To combat iron overload, iron chelation therapy is essential for all transfusion-dependent patients. Chelation agents, such as deferoxamine, deferasirox, or deferiprone, bind to the excess iron in the bloodstream, allowing it to be excreted from the body. This therapy is critical for preventing organ damage and improving long-term outcomes.
Hematopoietic Stem Cell Transplantation (HSCT)
HSCT is the only curative treatment for Diamond-Blackfan Anemia (DBA). It involves replacing the patient’s defective hematopoietic stem cells with healthy ones from a suitable donor. HSCT is typically recommended for patients who are transfusion-dependent and have a compatible donor, such as a matched sibling.
While it offers a cure, it is a high-risk procedure with potential complications, including graft-versus-host disease (GVHD), which is why it is not the first-line treatment for all patients.
The management of Diamond-Blackfan Anemia (DBA) requires a multidisciplinary approach involving hematologists, cardiologists, endocrinologists, and other specialists to address the anemia, monitor for complications like iron overload, and manage the associated malformations.
Prognosis and Long-Term Follow-up
The prognosis for Diamond-Blackfan anemia (DBA) has improved significantly over the past few decades with advances in treatment. However, it remains a chronic condition that requires lifelong medical management and careful monitoring for long-term complications.
Risk of Developing Myelodysplastic Syndrome (MDS) and Acute Myeloid Leukemia (AML)
This is a major long-term concern for patients with Diamond-Blackfan anemia (DBA). Studies have shown a significantly increased risk of developing these specific types of cancer later in life.
This risk is a primary reason why patients need continuous follow-up care, including regular blood tests and, in some cases, periodic bone marrow evaluations, to detect any early signs of a malignant transformation.
Effects of Chronic Anemia and Iron Overload
For patients who are transfusion-dependent, the long-term consequences of chronic anemia and the accumulation of excess iron are a significant threat. Iron overload can cause damage to vital organs over time, including:
- Heart: Iron deposition in the heart can lead to cardiomyopathy, or heart muscle disease, which can be life-threatening.
- Liver: It can cause liver fibrosis and cirrhosis, impairing liver function.
- Endocrine System: Iron overload can affect the pancreas and pituitary gland, leading to conditions like diabetes and hypothyroidism. This is why ongoing iron chelation therapy and monitoring of iron levels are absolutely essential for a good long-term outcome.
Long-term Management and Quality of Life
The follow-up care for a Diamond-Blackfan anemia (DBA) patient is a lifelong journey. It requires a multidisciplinary team, including a pediatric hematologist, cardiologist, endocrinologist, and other specialists as needed.
The focus is on maintaining a stable hemoglobin level, managing iron levels, and addressing any associated congenital malformations or developmental issues. With proper management and patient compliance, many individuals with Diamond-Blackfan anemia (DBA) are able to lead full and productive lives. Genetic counseling is also a crucial component of care for affected individuals and their families.
Conclusion
Diamond-Blackfan Anemia serves as a powerful model for understanding how fundamental cellular processes, such as ribosome biogenesis, can lead to complex and severe clinical disorders. Continued research into novel therapeutic targets and the underlying genetic mechanisms holds promise for improving the long-term prognosis for individuals with DBA.
Glossary of Medical Terms
- Anemia: A condition in which the blood doesn’t have enough healthy red blood cells.
- Aplasia: The failure of an organ or tissue to develop or to function normally.
- Erythropoiesis: The process of producing red blood cells.
- Haploinsufficiency: The condition that results when the loss of function of one copy of a gene is sufficient to cause a disease.
- Hematopoiesis: The process of creating new blood cells.
- Macrocytic Anemia: Anemia characterized by red blood cells that are larger than normal.
- Ribosomopathy: A disease caused by defects in the production or function of ribosomes.
- p53: A tumor suppressor protein that regulates cell growth and division.
- Reticulocyte: An immature red blood cell.
Frequently Asked Questions (FAQs)
Is Diamond-Blackfan Anemia a type of leukemia?
No, Diamond-Blackfan anemia (DBA) is not a type of leukemia, although it is a bone marrow failure syndrome that increases a person’s risk of developing leukemia and other cancers over their lifetime. It is a distinct condition characterized by a specific problem with red blood cell production.
Is Diamond-Blackfan anemia (DBA) a genetic disorder?
Yes, Diamond-Blackfan anemia (DBA) is a genetic disorder caused by mutations in specific genes, most often those that encode ribosomal proteins. It can be inherited from a parent or occur as a new (de novo) mutation.
What is the long-term prognosis for someone with DBA?
The prognosis for individuals with Diamond-Blackfan anemia (DBA) varies widely depending on their response to treatment and the development of complications like iron overload or malignancy. With appropriate management, many individuals can live into adulthood, but they require lifelong monitoring and care.
Can a bone marrow transplant cure DBA?
Yes, a bone marrow transplant (hematopoietic stem cell transplantation) can cure the hematologic manifestations of Diamond-Blackfan anemia (DBA). However, it is a high-risk procedure and is typically reserved for patients who are not responsive to other therapies or who have developed life-threatening complications.
How long can you live with Diamond-Blackfan anemia?
Diamond-Blackfan Anemia (DBA) was once considered a fatal childhood disease, modern treatments have significantly improved the prognosis and life expectancy. However, it’s not as simple as a single number, as the outlook depends heavily on the specific management and long-term complications.
Based on recent studies and data from registries, the overall survival for people with Diamond-Blackfan anemia (DBA) has improved dramatically. Research indicates that approximately 75% of people with Diamond-Blackfan anemia (DBA) are alive at age 50, though data is limited due to the rarity of the condition. The median survival has been reported to be around 38 to 56 years, with most deaths linked to long-term complications rather than the disease itself.
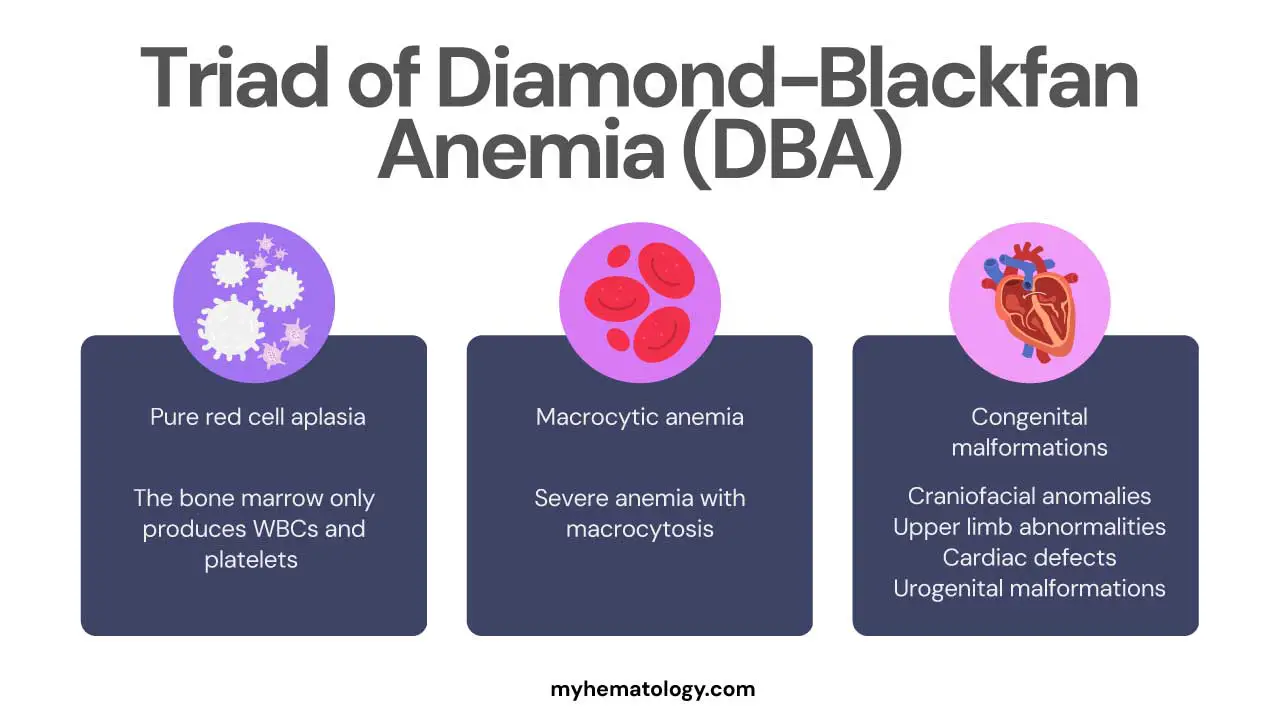
What is the triad of Diamond-Blackfan anemia?
There isn’t a single, universally accepted “triad” of symptoms for Diamond-Blackfan anemia (DBA). This term is often used clinically to refer to the three most prominent and diagnostic features of the condition:
- Pure Red Cell Aplasia: This is the most crucial hematological finding. The bone marrow selectively fails to produce red blood cells, while the other cell lines (white blood cells and platelets) remain normal.
- Macrocytic Anemia: The patient presents with a severe anemia where the red blood cells that are present are larger than normal.
- Congenital Malformations: The presence of associated physical anomalies, particularly those affecting the craniofacial region and upper limbs (like thumb abnormalities), is a hallmark of the disease and a key clue for diagnosis.
What is another name for Diamond-Blackfan anemia?
Another name for Diamond-Blackfan anemia is congenital hypoplastic anemia. This older name refers to the fact that the condition is present from birth (congenital) and is characterized by a reduced number of red blood cells due to a failure in bone marrow production (hypoplastic).
Disclaimer: This article is intended for informational purposes only and is specifically targeted towards medical students. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Killeen RB, Wills C. Diamond-Blackfan Anemia. [Updated 2025 Jul 7]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK545302/
- Engidaye, G., Melku, M., & Enawgaw, B. (2019). Diamond Blackfan Anemia: Genetics, Pathogenesis, Diagnosis and Treatment. EJIFCC, 30(1), 67–81.
- Da Costa, L. M., Marie, I., & Leblanc, T. M. (2021). Diamond-Blackfan anemia. Hematology. American Society of Hematology. Education Program, 2021(1), 353–360. https://doi.org/10.1182/hematology.2021000314
- Bhoopalan, S. V., Suryaprakash, S., Sharma, A., & Wlodarski, M. W. (2023). Hematopoietic cell transplantation and gene therapy for Diamond-Blackfan anemia: state of the art and science. Frontiers in oncology, 13, 1236038. https://doi.org/10.3389/fonc.2023.1236038
- Liu, Y., Dahl, M., Debnath, S., Rothe, M., Smith, E. M., Grahn, T. H. M., Warsi, S., Chen, J., Flygare, J., Schambach, A., & Karlsson, S. (2022). Successful gene therapy of Diamond-Blackfan anemia in a mouse model and human CD34+ cord blood hematopoietic stem cells using a clinically applicable lentiviral vector. Haematologica, 107(2), 446–456. https://doi.org/10.3324/haematol.2020.269142
- Liu, Y., Karlsson, S. Perspectives of current understanding and therapeutics of Diamond-Blackfan anemia. Leukemia 38, 1–9 (2024). https://doi.org/10.1038/s41375-023-02082-w
- Aspesi, A., Borsotti, C., & Follenzi, A. (2018). Emerging Therapeutic Approaches for Diamond Blackfan Anemia. Current gene therapy, 18(6), 327–335. https://doi.org/10.2174/1566523218666181109124538