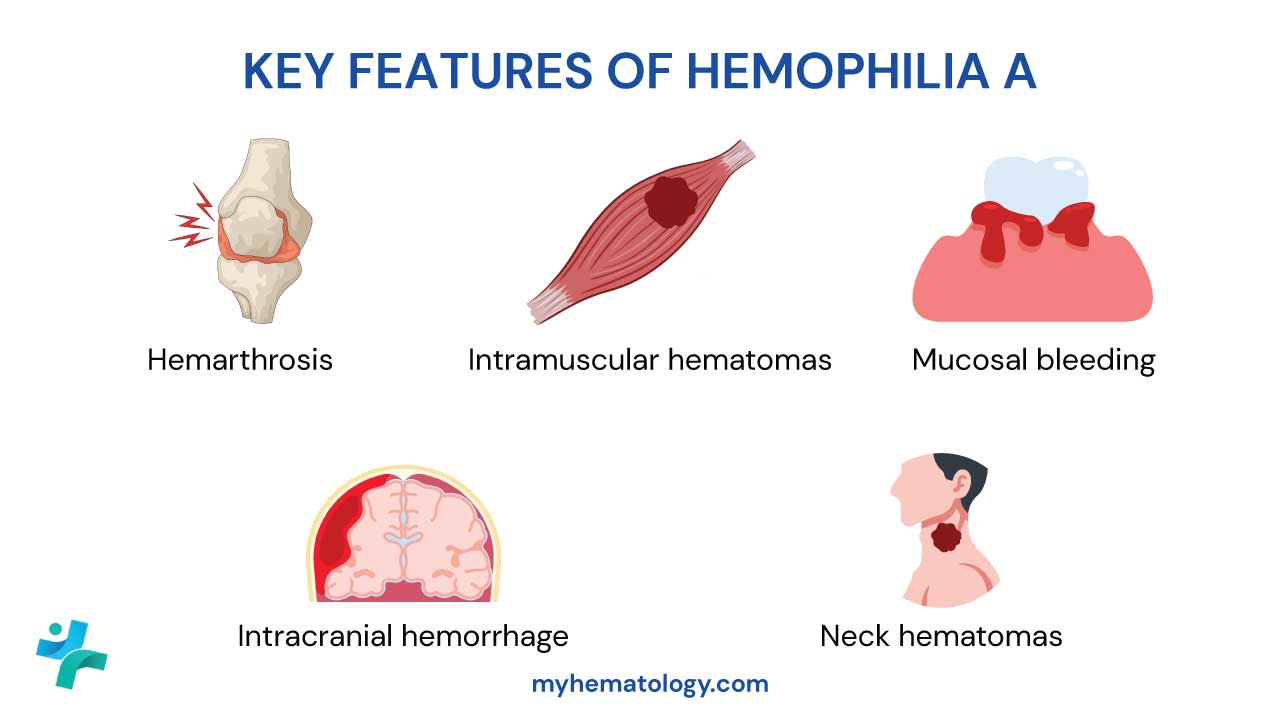
Key Takeaways
Von Willebrand disorder, also called von Willebrand disease (VWD), is the most common inherited bleeding disorder, affecting roughly 1 in 1,000 people, and is caused by a missing or defective protein called von Willebrand factor that normally helps platelets stick to wounds and protects clotting factor VIII [3].
Pathogenesis ▾: VWD has three main types: type 1 (partial quantitative deficiency, most common, mild, autosomal dominant), type 2 (qualitative defect with subtypes 2A, 2B, 2M, 2N), and type 3 (complete deficiency, rare, severe, autosomal recessive); a separate "low VWF" category covers VWF levels of 30–50 IU/dL [1].
Signs and symptoms ▾: Excessive bleeding from cuts, injuries, surgery, dental procedures, and menstruation or easy bruising and frequent nosebleeds.
Laboratory diagnosis ▾: Diagnosis of von Willebrand disorder begins with a Bleeding Assessment Tool, then measures VWF antigen, VWF activity (often by VWF:GPIbM or GPIbR assays), and factor VIII activity, with multimer analysis, RIPA, and genetic testing reserved for subtyping [1].
Treatment and management ▾: First-line treatment for mild type 1 VWD is desmopressin (DDAVP), while VWF concentrates (plasma-derived or recombinant) are essential for type 3, type 2B, and major surgery; tranexamic acid and hormonal therapy support bleeding control in dental procedures and heavy periods [2].
*Click ▾ for more information
Von Willebrand disorder is the most common inherited bleeding problem in the world, yet many people have never heard of it. If you have a friend who bruises after the lightest knock, a relative whose periods last ten days, or a patient on the ward whose nosebleeds will not stop, von Willebrand disorder belongs on the list of suspects. This article walks through what it is, why it causes bleeding, how it is diagnosed, and how it is treated, using the current 2021 international guidelines as a backbone [1,2].
von Willebrand disorder and von Willebrand disease refer to the same condition and are used interchangeably. The standard abbreviation is VWD.
What is von Willebrand Disorder or VWD?
Von Willebrand disorder is a bleeding condition caused by a missing or faulty protein called von Willebrand factor (VWF). It affects about 1 in 1,000 people worldwide, although only a fraction are formally diagnosed [3]. The Finnish physician Erik von Willebrand first described it in 1926, in a young girl from the Åland Islands whose family bled heavily across generations. While population screening suggests up to 1% of the general population carries low VWF levels or a genetic variant, only about 1 in 1,000 to 1 in 10,000 people experience symptomatic bleeding severe enough to require medical attention [11].
Older textbooks list higher prevalence in North America and Europe than in sub-Saharan Africa or parts of Asia. This pattern mostly reflects who has access to specialized testing, not true biological differences. Wherever populations are screened thoroughly, VWD turns up.
VWD is usually inherited, but a smaller number of people develop a similar condition later in life called acquired von Willebrand syndrome, which we cover in a dedicated section below.
What is von Willebrand Factor or VWF?
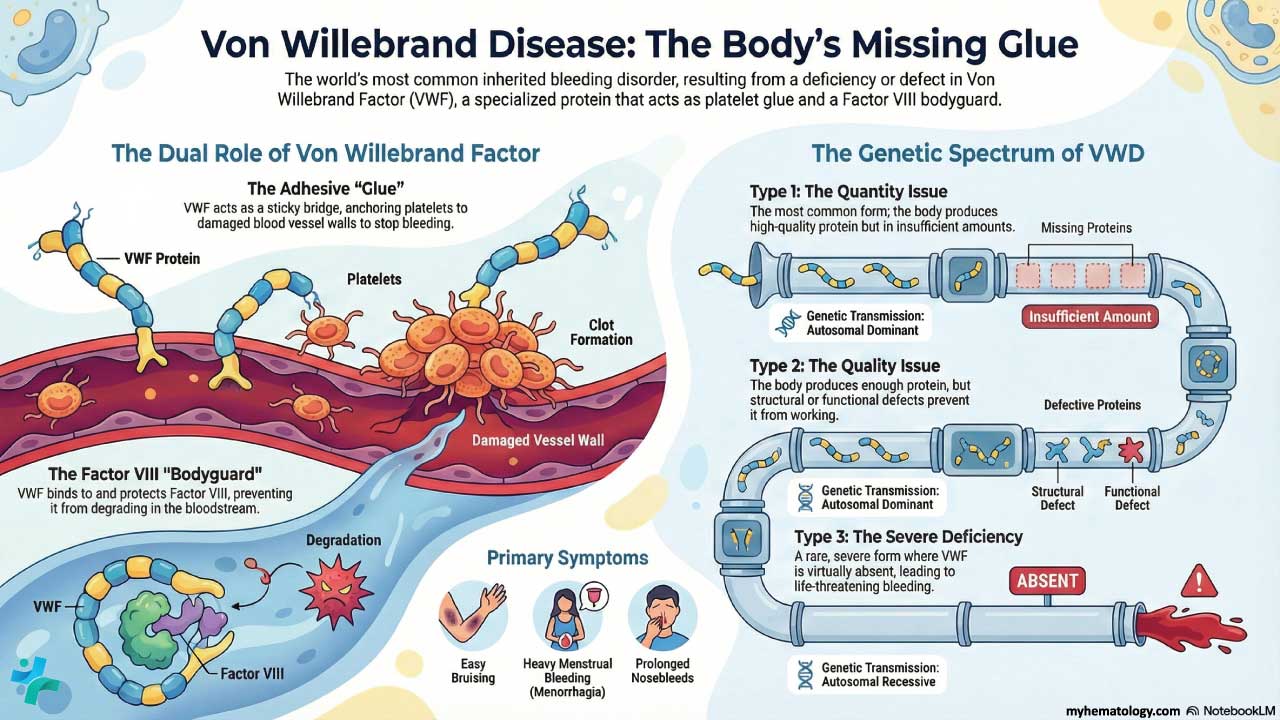
VWF is a large glycoprotein (a sugar-coated protein) made by two cell types: the endothelial cells lining your blood vessels, and the megakaryocytes in bone marrow that produce platelets. The gene that codes for it sits on chromosome 12 (band 12p13.31), spans roughly 178 kilobases, and contains 52 exons.
Once secreted into plasma, VWF molecules join together into long chains called multimers. The biggest multimers are the most active. They have two essential jobs in hemostasis — the process of stopping bleeding [3,5]:
- Holding platelets to wounded vessel walls. When a vessel is cut, VWF acts like double-sided tape, anchoring platelets to exposed collagen so a plug can form.
- Protecting clotting factor VIII (FVIII). VWF binds FVIII in the bloodstream and shields it from being broken down. Without VWF, FVIII levels drop.
That second job is why VWD can look surprisingly like hemophilia A in its severe forms. The platelet-plug step fails and the fibrin-clot step is weakened.
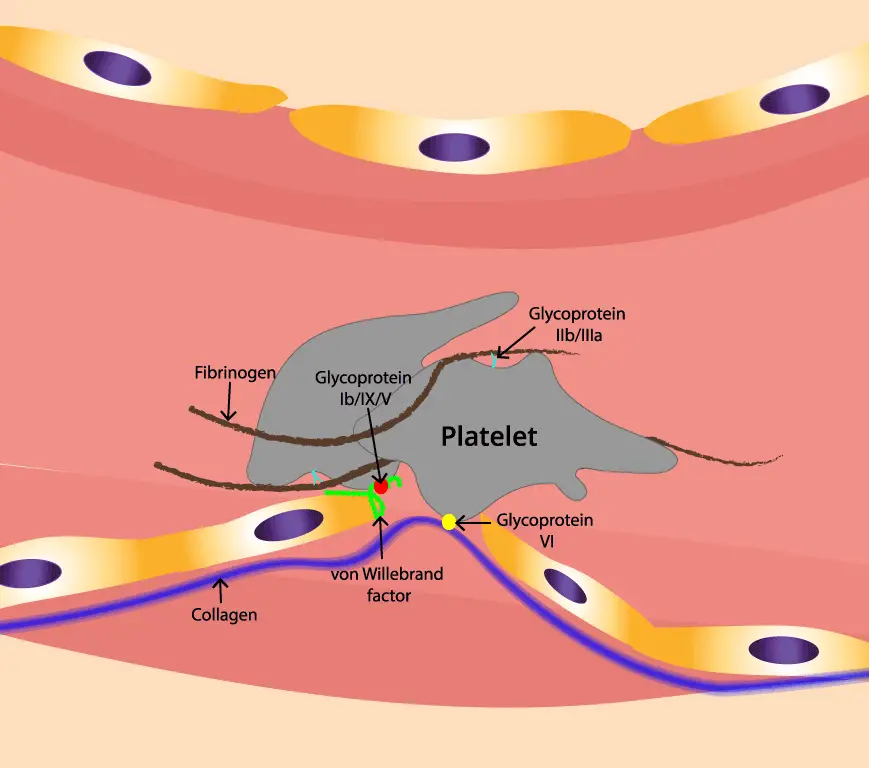
vWD Types
VWD is divided into three main types based on whether VWF is missing in quantity or defective in quality [1,6].
Type 1 — partial quantitative deficiency. The most common form (roughly 70% of cases). VWF works normally but there is not enough of it. Inherited as autosomal dominant. Symptoms are usually mild.
Type 2 — qualitative defect. About 20–25% of cases. The amount of VWF may be normal, but the protein does not function properly. Type 2 is split into four subtypes:
- Type 2A: large multimers fail to form or are cleared too quickly. Most common type 2 subtype.
- Type 2B: VWF binds platelets too aggressively, leading to loss of large multimers and sometimes a low platelet count (thrombocytopenia, meaning fewer platelets than normal).
- Type 2M: VWF cannot bind platelets properly, but multimer assembly is normal.
- Type 2N (Normandy): VWF cannot bind FVIII, so FVIII levels drop. Looks clinically like mild hemophilia A. Inherited as autosomal recessive.
Type 3 — complete deficiency. The rarest form, around 5%. VWF is essentially absent. Inherited as autosomal recessive. Bleeding is severe and can include hemarthrosis (bleeding into joints).
A separate category called "low VWF" has emerged in current guidelines [1]. People with VWF levels of 30–50 IU/dL no longer meet the strict cutoff for type 1 VWD (which is now <30 IU/dL) but may still bleed and need management.

How does von Willebrand disorder cause bleeding?
VWD interrupts both stages of hemostasis at once.
Primary hemostasis fails because platelets cannot stick to the vessel wall without enough functional VWF. This produces the classic mucocutaneous bleeding pattern: bleeding from skin and mucous membranes — easy bruising, gum bleeds, nosebleeds, heavy periods, and oozing after dental work [3].
Secondary hemostasis weakens because FVIII has lost its carrier and gets degraded faster. When FVIII drops low enough (typically in type 3 and type 2N) bleeding extends into joints and muscles, mimicking hemophilia A.
This dual failure also explains the long-term complications: chronic blood loss can cause iron deficiency anemia (low red blood cell count from depleted iron stores), repeated joint bleeds can cause arthritis, and pregnancy carries a higher risk of postpartum hemorrhage.
Signs and Symptoms
Symptoms vary widely. Mild type 1 may go unnoticed until a tooth extraction or first period; type 3 announces itself in infancy.
Common, mild symptoms (usually type 1):
- Easy or frequent bruising, sometimes with lumps under the skin
- Frequent nosebleeds (≥5 per year, or lasting more than 10 minutes)
- Bleeding gums after brushing
- Prolonged bleeding from minor cuts and after dental procedures or surgery
Symptoms that disproportionately affect women:
- Heavy or prolonged menstrual periods (menorrhagia) — soaking through a pad or tampon every 1–2 hours, periods longer than 7 days, or clots larger than a grape
- Iron deficiency anemia from chronic blood loss, causing fatigue and breathlessness
- Heavy bleeding after childbirth or miscarriage
Severe symptoms (mostly type 3, occasionally type 2N):
- Spontaneous bleeding with no clear trigger
- Hemarthrosis (joint bleeds) causing swelling, stiffness, and pain
- Gastrointestinal bleeding, with blood in the stool
- Hematuria (blood in the urine)
How is Von Willebrand disorder diagnosed?
Diagnosis follows a structured, multi-step process. The 2021 international guidelines [1] recommend starting with a Bleeding Assessment Tool (BAT) — a scored questionnaire (for example, the ISTH-BAT or Self-BAT) that turns "do you bleed a lot?" into a quantifiable number. Patients with elevated bleeding scores then move to laboratory testing.
Routine screening tests
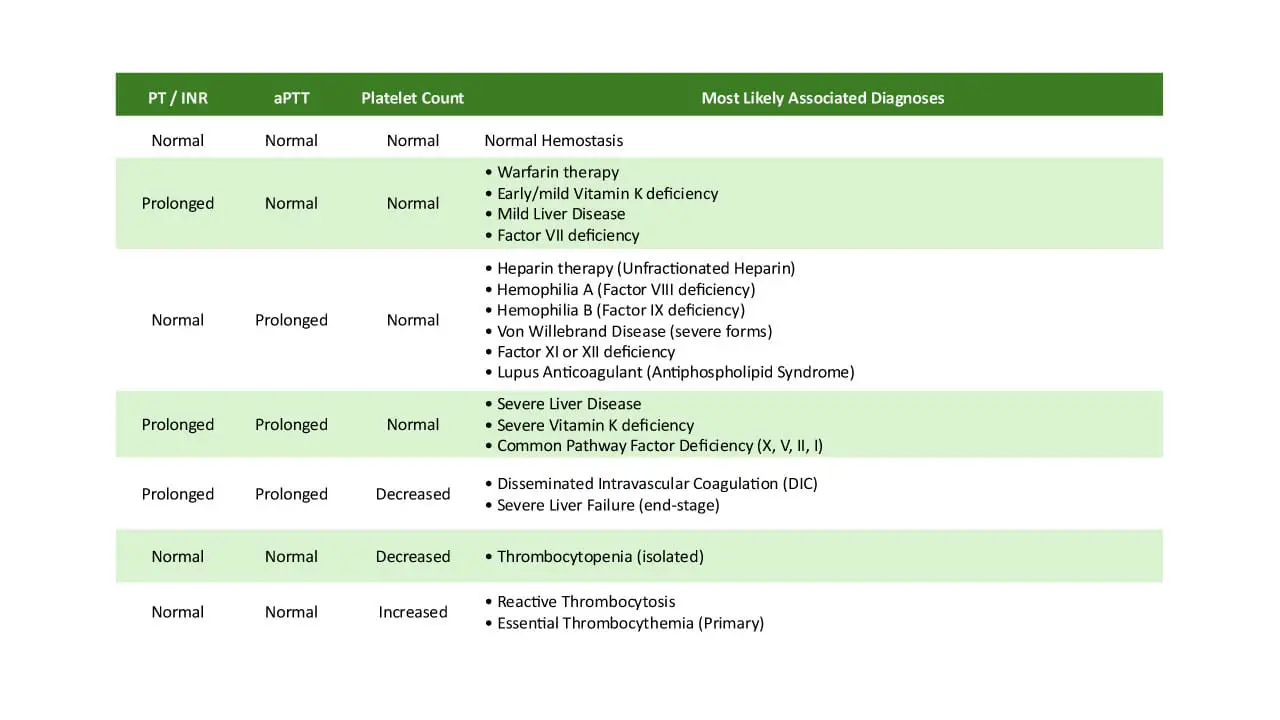
These initial coagulation tests are often normal in mild VWD but are part of any bleeding workup:
- Complete blood count (CBC): platelet count is usually normal, except in type 2B where it can be low.
- Prothrombin time (PT): normal in all VWD subtypes.
- Activated partial thromboplastin time (aPTT): normal or mildly prolonged, with stronger prolongation when FVIII is very low (type 3, type 2N).
| Test name | What it measures | Clinical significance in VWD |
|---|---|---|
| VWF antigen (VWF:Ag) | Total amount of VWF in plasma | Low in type 1 and type 3 |
| VWF activity (VWF:Act) | How well VWF binds platelets | Low activity with normal or near-normal antigen suggests type 2 |
| FVIII coagulant activity (FVIII:C) | The activity of clotting factor VIII, which VWF stabilizes and protects from degradation | Often low, especially in type 3 and type 2N |
Modern labs increasingly use VWF:GPIbM or VWF:GPIbR assays in place of the older VWF:RCo (ristocetin cofactor) test, because the GPIb-based assays are more reproducible at low levels [1,4]. This transition to modern assays is critical because the older VWF:RCo test is notoriously prone to false-positive results in individuals with certain benign genetic variations (most notably in populations of African descent) and often lacks diagnostic accuracy at lower VWF concentrations [12].
Tier 2: Identifying the Subtype
If Tier 1 is abnormal, the next step is to pin down which subtype. This is essential because treatment differs.
- VWF multimer analysis uses gel electrophoresis to visualize VWF chain sizes. Loss of high-molecular-weight multimers points to type 2A or 2B.
- Ristocetin-induced platelet aggregation (RIPA) tests platelet–VWF binding in the presence of the antibiotic ristocetin. Increased aggregation at low ristocetin doses is the fingerprint of type 2B.
- VWF collagen-binding assay (VWF:CBA) flags type 2A.
- VWF–FVIII binding assay (VWF:FVIIIB) confirms type 2N and distinguishes it from mild hemophilia A.
- Genetic testing of the VWF gene is reserved for confirming type 2N and type 3, resolving ambiguous results, and family planning.
Laboratory Reference
| VWD Type | VWF:Ag | VWF:Act | FVIII:C | Activity / Antigen Ratio | Multimer Analysis |
|---|---|---|---|---|---|
|
Normal
|
50–200 IU/dL | N | N | ≈ 1.0 | Full range |
|
Type 1
|
↓ proportionally | ↓ proportionally | ↓ or N | ≥ 0.7 | Normal |
|
Type 2A
|
N or ↓ | ↓↓ | N or ↓ | < 0.7 | HMW absent |
|
Type 2B
|
N or ↓ | ↓↓ | N or ↓ | < 0.7 | HMW absent |
|
Type 2M
|
N or ↓ | ↓↓ | N or ↓ | < 0.7 | Normal |
|
Type 2N
|
N or ↓ | N or ↓ | ↓↓ | ≥ 0.7 | Normal |
|
Type 3
|
Absent | Absent | ↓↓ (Severe) | N/A | Absent |
Differential Diagnosis of VWD
| Condition | Mechanism | Inheritance | Bleeding Pattern | aPTT | FVIII:C | VWF:Ag / Act | Platelet Count | |
|---|---|---|---|---|---|---|---|---|
|
Von Willebrand Disease (VWD)
Types 1, 2A, 2B, 2M, 2N, 3
|
VWF deficient or dysfunctional |
AD — Types 1, 2A, 2B, 2M
AR — Types 3, 2N
|
Mucocutaneous; deep tissue in severe cases | Normal (Type 1, 2) to Prolonged (Type 3, 2N) | Normal to ↓↓ | ↓ | Usually normal (low in Type 2B) | |
|
Hemophilia A
Classic hemophilia
|
FVIII deficiency | X-linked Recessive | Primarily deep tissue bleeding; haemarthrosis | Prolonged | ↓↓ | Normal | Normal | |
| Platelet Function Disorders | Defect in platelet aggregation | Autosomal Recessive | Mucocutaneous, often severe | Normal | Normal | Normal | Normal count, abnormal function | |
| Thrombocytopenia | Low platelet number | Variable | Petechiae, purpura, mucocutaneous | Normal | Normal | Normal | Low | |
|
Type 2N VWD
VWF–FVIII binding defect
|
VWF cannot bind FVIII | Autosomal Recessive | Like mild Haemophilia A | Prolonged | ↓↓ | Normal — VWF:FVIIIB is the giveaway | Normal |
How is VWD treated?
Treatment of von Willebrand disorder depends on the subtype, the severity, and the situation — managing a current bleed, preventing one before surgery, or controlling heavy periods. The aim is to raise functional VWF (and, by extension, FVIII) to a level that supports normal clotting [2].
Primary treatment agents
Desmopressin (DDAVP). A synthetic hormone that prompts endothelial cells to release their stored VWF and FVIII. Given intravenously, subcutaneously, or as a nasal spray. It is first-line for most type 1 patients and may work in some type 2A or 2M cases but only after a trial dose confirms response. DDAVP is contraindicated in type 2B because releasing more abnormal VWF triggers platelet binding and can drop the platelet count further [2].
When utilizing DDAVP, clinicians must navigate several strict safety caveats. It is contraindicated in children under 2 years of age due to a high risk of severe hyponatremia (low sodium) and subsequent seizures, and it must be used with extreme caution in elderly patients with cardiovascular disease. Patients require strict fluid intake restriction for 24 hours post-dose. Furthermore, repeated dosing over a short period leads to tachyphylaxis—a diminished clinical response as the endothelial storage pools of VWF become depleted [13].
VWF and Factor VIII concentrates. Infused directly into a vein.
- Plasma-derived concentrates (pd-VWF/FVIII) contain both VWF and FVIII and undergo pathogen-reduction steps such as solvent/detergent treatment, nanofiltration, and heat to minimize infection risk.
- Recombinant VWF (rVWF, vonicog alfa) is genetically engineered, contains no human plasma, and allows independent dosing of VWF without simultaneously raising FVIII.
Concentrates are essential for type 3, the treatment of choice for type 2B (where DDAVP is unsafe), and for major surgery or trauma in any subtype. Current guideline-recommended VWF activity targets are roughly >50 IU/dL for minor procedures and >100 IU/dL for major surgery [2].
Routine prophylaxis
Historically, VWD treatment was largely on-demand, but clinical practice has shifted strongly toward routine prophylaxis (preventative treatment) for patients with severe phenotypes, such as type 3 and severe forms of types 1 and 2. This proactive approach significantly reduces spontaneous bleeds, prevents irreversible joint damage, and improves quality of life. Recent regulatory milestones have solidified this standard of care, including the 2023 FDA approval of the plasma-derived VWF/FVIII concentrate Wilate for routine prophylaxis in adults and children, alongside the expanded prophylactic use of recombinant VWF (vonicog alfa) for severe VWD [14,15].
These medications help stabilize the clot once it has formed and are often used in combination with DDAVP or concentrates.
| Drug Class | Examples | Action | Common Use |
|---|---|---|---|
|
Antifibrinolytics
Hemostatic support
|
Tranexamic acid (TXA), Aminocaproic acid (Amicar) | Stop clots from being broken down too quickly |
|
|
Hormonal Therapy
VWF modulation
|
Combined oral contraceptives, levonorgestrel IUD | Estrogen raises endogenous VWF and FVIII; progestin thins the endometrium | |
|
Topical Agents
Local hemostasis
|
Fibrin Sealant, Thrombin Spray | Promote local clotting at the bleeding site |
|
Pregnancy and delivery
Most women with VWD have safe pregnancies. VWF levels rise naturally during the third trimester, especially in type 1, and often reach normal range by delivery. The 2021 guidelines recommend [2]:
- Target VWF:Act >50 IU/dL at the time of delivery
- Neuraxial anesthesia (epidural/spinal) is acceptable when VWF:Act is >50 IU/dL
- Tranexamic acid as first-line for postpartum hemorrhage prevention
- Care coordinated between a hematologist, obstetrician, and anesthesiologist
Postpartum hemorrhage risk remains elevated for several weeks after delivery as VWF levels fall back, and warrants monitoring.
General management
- Avoid antiplatelet drugs. Aspirin, ibuprofen, and naproxen further impair platelet function. Acetaminophen (paracetamol) is generally safer for pain and fever.
- Plan elective procedures. Any surgery, including dental work, needs a hematologist-led plan with target factor levels and a treatment schedule.
- Use specialist centers. Hemophilia Treatment Centers (HTCs) provide multidisciplinary care that improves outcomes.
- Carry medical identification. A bracelet or alert card ensures appropriate emergency treatment.
Acquired von Willebrand syndrome (AVWS)
Not all VWF deficiency is inherited. Acquired von Willebrand syndrome develops later in life from another disease process. Common triggers include monoclonal gammopathies and lymphoproliferative disorders, severe aortic stenosis (Heyde syndrome), myeloproliferative neoplasms such as essential thrombocythemia, hypothyroidism, and mechanical circulatory support (LVADs) [9]. AVWS is underrecognized in older patients. Symptoms mirror inherited VWD but resolve when the underlying cause is treated. Treatment in the meantime relies on DDAVP, IVIG (in monoclonal gammopathy–related cases), VWF concentrates, and antifibrinolytics.
Emerging therapies
The treatment landscape is broadening [4,7,8]. Recombinant VWF is already in routine use. Novel agents in clinical development include:
- KB-V13A12, a nanobody-based therapeutic designed to extend VWF half-life
- HMB-002, a monovalent antibody binding endogenous VWF and entering phase I trials in type 1 VWD in 2025
- Rondoraptivon pegol (BT200), a pegylated RNA aptamer originally developed as an antithrombotic but showing potential in VWD
- VGA039, a highly anticipated subcutaneous monoclonal antibody targeting protein S to enhance thrombin generation. Currently advancing through late-stage clinical trials, it is positioned as a potential first-in-class under-the-skin prophylactic option for VWD [16].
- Emicizumab, while already established in hemophilia A care, this subcutaneous FVIII-mimicking bispecific antibody is under active clinical investigation for VWD, specifically targeting patients with severe secondary FVIII deficiency (such as type 3) [17].
- Efanesoctocog alfa (BIVV-001), a novel extended half-life factor VIII therapy currently in clinical trials to evaluate its efficacy in VWD types 2N and 3 [18].
Gene therapy for VWD has lagged behind hemophilia gene therapy because the VWF cDNA is large (8 kb) and difficult to package into standard viral vectors. For now, careful use of existing therapies controls bleeding effectively in most patients.
When to seek urgent care
Anyone with VWD and the people who care for them should know which bleeding events demand immediate attention:
- A nosebleed lasting more than 30 minutes despite firm pinching
- Bleeding that soaks through dressings or bandages
- Vomiting or coughing up blood, or passing black, tarry stools
- A severe headache, confusion, or new neurological symptoms after a head injury
- Heavy menstrual bleeding causing dizziness or fainting
- Joint swelling and pain after a knock, especially in known type 3 patients
Frequently Asked Questions (FAQs)
What is von Willebrand disorder in simple terms?
Von Willebrand disorder, also called von Willebrand disease (VWD), is the most common inherited bleeding disorder, affecting roughly 1 in 1,000 people. It is caused by a missing or faulty protein called von Willebrand factor (VWF), which normally helps platelets stick to injured blood vessels and protects clotting factor VIII. People with VWD bruise easily, get nosebleeds, bleed longer after cuts or surgery, and women often have heavy menstrual periods. Most cases are mild and manageable.
How is von Willebrand disorder diagnosed?
Diagnosis of von Willebrand disorder begins with a bleeding history (often scored with a Bleeding Assessment Tool such as the ISTH-BAT) and a physical exam. If bleeding is unusual for the person's age and sex, blood tests measure VWF antigen (how much VWF is present), VWF activity (how well it works), and factor VIII activity. Under current 2021 international guidelines, a VWF level below 30 IU/dL confirms VWD, while levels of 30–50 IU/dL are classified as "low VWF." Further tests, including multimer analysis and genetic testing, identify the specific subtype.
Is von Willebrand disorder dangerous?
For most people, von Willebrand disorder is not life-threatening. Type 1, which is the mildest and most common form (about 70% of cases), usually causes only manageable symptoms such as easy bruising and heavy periods. Type 2 and especially type 3 carry a higher risk of serious bleeding from surgery, childbirth, injuries, or sometimes spontaneously into joints. With proper diagnosis and a treatment plan from a hematologist, life expectancy is normal.
How is von Willebrand disorder treated?
Treatment of von Willebrand disorder is tailored to the subtype and the bleeding situation. For mild type 1, desmopressin (DDAVP) is first-line — it triggers release of the body's stored VWF. For type 3, type 2B, and major surgery, VWF concentrates (plasma-derived or recombinant) replace the missing protein. Antifibrinolytic drugs like tranexamic acid help stop bleeding from the mouth, nose, and uterus. Hormonal therapy such as combined oral contraceptives is often used to control heavy periods. Patients should avoid aspirin and NSAIDs unless cleared by their hematologist.
Can people with von Willebrand disorder have children safely?
Yes. Most women with von Willebrand disorder have safe pregnancies and healthy babies, especially with type 1, where VWF levels naturally rise during pregnancy. Care should be coordinated by a hematologist and obstetrician. The current goal is a VWF activity level above 50 IU/dL at delivery; tranexamic acid and VWF concentrates are used when needed. Postpartum hemorrhage risk remains elevated for several weeks after delivery and warrants monitoring. Each child has up to a 50% chance of inheriting dominant forms of VWD.
What is the difference between inherited VWD and acquired von Willebrand syndrome?
Inherited von Willebrand disorder is present from birth due to a mutation in the VWF gene. Acquired von Willebrand syndrome (AVWS) develops later in life as a complication of another condition — most often monoclonal gammopathy, aortic valve stenosis (Heyde syndrome), certain cancers like lymphoma, or mechanical heart pumps (LVADs). AVWS produces similar bleeding symptoms but resolves when the underlying cause is treated.
Glossary of Related Medical Terms
- Hemostasis — The body's process of stopping bleeding after a blood vessel is damaged.
- Primary hemostasis — The first response, in which platelets stick to the injury site and form a temporary plug.
- Secondary hemostasis — The follow-up step, in which clotting factors form a fibrin mesh that stabilizes the platelet plug.
- Von Willebrand factor (VWF) — A large protein in the blood that helps platelets stick to a wounded vessel and protects clotting factor VIII from being broken down.
- Multimer — A long chain made of many copies of the same protein joined together. Larger VWF multimers work better at trapping platelets.
- Endothelial cells — The thin layer of cells that lines the inside of every blood vessel.
- Megakaryocytes — Bone marrow cells that pinch off platelets into the blood.
- Platelet adhesion — Platelets sticking to a damaged vessel wall, the first step in plug formation.
- Mucocutaneous bleeding — Bleeding from skin and mucous membranes (gums, nose, gut, uterus). Typical of VWD.
- Hemarthrosis — Bleeding into a joint, causing pain and swelling. Common in severe VWD and hemophilia.
- Menorrhagia — Heavy or prolonged menstrual bleeding.
- Epistaxis — Nosebleed.
- Thrombocytopenia — Low platelet count.
- Antigen (in lab tests) — A measurement of how much of a protein is present, regardless of whether it works.
- Activity (in lab tests) — A measurement of how well a protein performs its job.
- Ristocetin — An antibiotic that triggers VWF–platelet binding in lab tests; used to measure VWF function.
- Desmopressin (DDAVP) — A synthetic hormone that prompts the body to release stored VWF and FVIII.
- Antifibrinolytic — A drug (such as tranexamic acid) that prevents clots from being broken down too quickly.
- Acquired von Willebrand syndrome (AVWS) — A non-inherited form of VWF deficiency caused by another medical condition.
- Bleeding Assessment Tool (BAT) — A scored questionnaire that estimates how unusual a person's bleeding history is, used to decide who needs lab testing.
Disclaimer: This article is intended for educational and informational purposes only. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- James, P. D., Connell, N. T., Ameer, B., Di Paola, J., Eikenboom, J., Giraud, N., Haberichter, S., Jacobs-Pratt, V., Konkle, B., McLintock, C., McRae, S., R Montgomery, R., O'Donnell, J. S., Scappe, N., Sidonio, R., Flood, V. H., Husainat, N., Kalot, M. A., & Mustafa, R. A. (2021). ASH ISTH NHF WFH 2021 guidelines on the diagnosis of von Willebrand disease. Blood advances, 5(1), 280–300. https://doi.org/10.1182/bloodadvances.2020003265
- Connell, N. T., Flood, V. H., Brignardello-Petersen, R., Abdul-Kadir, R., Arapshian, A., Couper, S., Grow, J. M., Kouides, P., Laffan, M., Lavin, M., Leebeek, F. W. G., O'Brien, S. H., Ozelo, M. C., Tosetto, A., Weyand, A. C., James, P. D., Kalot, M. A., Husainat, N., & Mustafa, R. A. (2021). ASH ISTH NHF WFH 2021 guidelines on the management of von Willebrand disease. Blood advances, 5(1), 301–325. https://doi.org/10.1182/bloodadvances.2020003264
- Seidizadeh, O., Eikenboom, J. C. J., Denis, C. V., Flood, V. H., James, P., Lenting, P. J., Baronciani, L., O'Donnell, J. S., Lillicrap, D., & Peyvandi, F. (2024). von Willebrand disease. Nature reviews. Disease primers, 10(1), 51. https://doi.org/10.1038/s41572-024-00536-8
- Connell N. T. (2026). Treatment of von Willebrand disease. Blood advances, 10(3), 794–801. https://doi.org/10.1182/bloodadvances.2025016487
- Leebeek, F. W., & Eikenboom, J. C. (2016). Von Willebrand's Disease. The New England journal of medicine, 375(21), 2067–2080. https://doi.org/10.1056/NEJMra1601561
- Sadler, J. E., Budde, U., Eikenboom, J. C., Favaloro, E. J., Hill, F. G., Holmberg, L., Ingerslev, J., Lee, C. A., Lillicrap, D., Mannucci, P. M., Mazurier, C., Meyer, D., Nichols, W. L., Nishino, M., Peake, I. R., Rodeghiero, F., Schneppenheim, R., Ruggeri, Z. M., Srivastava, A., Montgomery, R. R., … Working Party on von Willebrand Disease Classification (2006). Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. Journal of thrombosis and haemostasis : JTH, 4(10), 2103–2114. https://doi.org/10.1111/j.1538-7836.2006.02146.x
- Favaloro, E. J., Pasalic, L., & Curnow, J. (2024). Current and emerging therapies as potential treatment for people with von Willebrand disease. Expert review of hematology, 17(12), 917–933. https://doi.org/10.1080/17474086.2024.2429611
- Casari, C., Leebeek, F. W. G., & Peyvandi, F. (2026). Historical, current and future treatments for von Willebrand disease. Haematologica, 111(1), 54–66. https://doi.org/10.3324/haematol.2024.286037
- Leebeek F. W. G. (2021). New Developments in Diagnosis and Management of Acquired Hemophilia and Acquired von Willebrand Syndrome. HemaSphere, 5(6), e586. https://doi.org/10.1097/HS9.0000000000000586
- Sabih A, Babiker HM. Von Willebrand Disease. [Updated 2023 Aug 28]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2026 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK459222/
- Bowman, M., Hopman, W. M., Rapson, D., Lillicrap, D., & James, P. (2010). The prevalence of symptomatic von Willebrand disease in primary care practice. Journal of thrombosis and haemostasis : JTH, 8(1), 213–216. https://doi.org/10.1111/j.1538-7836.2009.03661.x
- Flood, V. H., Gill, J. C., Morateck, P. A., Christopherson, P. A., Friedman, K. D., Haberichter, S. L., Branchford, B. R., Hoffmann, R. G., Abshire, T. C., Di Paola, J. A., Hoots, W. K., Leissinger, C., Lusher, J. M., Ragni, M. V., Shapiro, A. D., & Montgomery, R. R. (2010). Common VWF exon 28 polymorphisms in African Americans affecting the VWF activity assay by ristocetin cofactor. Blood, 116(2), 280–286. https://doi.org/10.1182/blood-2009-10-249102
- Leissinger, C., Carcao, M., Gill, J.C., Journeycake, J., Singleton, T. and Valentino, L. (2014), Desmopressin (DDAVP) in the management of patients with congenital bleeding disorders. Haemophilia, 20: 158-167. https://doi.org/10.1111/hae.12254
- U.S. Food and Drug Administration. (2023). FDA approves Wilate for routine prophylaxis in adults and children with von Willebrand disease. FDA.gov.
- van Kwawegen, C. B., & Leebeek, F. W. G. (2024). Prophylaxis in von Willebrand disease with von Willebrand factor concentrate and nonfactor therapies. Research and practice in thrombosis and haemostasis, 8(8), 102599. https://doi.org/10.1016/j.rpth.2024.102599
- Prince Eladnani R, Diab R, Angelillo-Scherrer A. (2025). Protein S as a therapeutic target. Journal of Thrombosis and Haemostasis, 24, 354-367. https://doi.org/10.1016/j.jtha.2025.10.024
- Swan, D., Mahlangu, J., & Thachil, J. (2022). Non-factor therapies for bleeding disorders: A primer for the general haematologist. EJHaem, 3(3), 584–595. https://doi.org/10.1002/jha2.442
- Dargaud, Y., Leuci, A., Ruiz, A. R., & Lacroix-Desmazes, S. (2024). Efanesoctocog alfa: the renaissance of Factor VIII replacement therapy. Haematologica, 109(8), 2436–2444. https://doi.org/10.3324/haematol.2023.284498