Key Takeaways
Langerhans Cell Histiocytosis or formerly known as Histiocytosis X is a clonal myeloid neoplasm characterized by the proliferation of abnormal cells resembling Langerhans cells. It is driven by inflammatory signaling rather than simple cell mass.
- Molecular Driver ▾: Over 80% of cases involve mutations in the MAPK/ERK pathway, most commonly the BRAFV600E mutation (~50%).
- Clinical Hallmark ▾: The "great mimicker," presenting as anything from a solitary "punched-out" lytic bone lesion to multisystem organ failure.
- Risk Stratification ▾: Prognosis is dictated by the involvement of Risk Organs: the Liver, Spleen, and Hematopoietic system (Bone Marrow).
- Diagnostic Gold Standard ▾: Tissue biopsy showing positive immunohistochemistry (IHC) for CD1a, S100, and Langerin (CD207).
- Treatment ▾:
- A 12-month regimen of Vinblastine and Prednisone for multisystem or high-risk disease.
- The emergence of targeted therapy (e.g., BRAF inhibitors like Vemurafenib) for refractory or high-risk cases.
*Click ▾ for more information
Introduction
Langerhans Cell Histiocytosis or formerly known as Histiocytosis X is a rare and clinically diverse disorder characterized by the clonal proliferation of pathological cells that closely resemble skin Langerhans cells. While historically debated as an inflammatory or reactive process, modern molecular medicine has redefined Langerhans Cell Histiocytosis (Histiocytosis X) as an inflammatory myeloid neoplasm.
Definition of Langerhans Cell Histiocytosis (Histiocytosis X)
At its core, Langerhans Cell Histiocytosis (Histiocytosis X) involves the accumulation of abnormal cells within various organs, most commonly the bone, skin, and pituitary gland. These cells trigger a robust local inflammatory response, leading to the formation of granulomas that cause tissue destruction.
According to the 2022 WHO Classification of Haematolymphoid Tumours, Langerhans Cell Histiocytosis (Histiocytosis X) is categorized under "Histiocytic and dendritic cell neoplasms." This classification emphasizes that despite the name, the disease originates from myeloid progenitor cells in the bone marrow rather than mature dendritic cells in the skin.
The "Myeloid Neoplasm" Paradigm
The defining feature of modern Langerhans Cell Histiocytosis (Histiocytosis X) education is the shift from "unknown cause" to "driver mutation."
- Clonality: In over 80% of cases, Langerhans Cell Histiocytosis (Histiocytosis X) cells harbor somatic mutations in the MAPK (Mitogen-Activated Protein Kinase) signaling pathway.
- The BRAFV600E Mutation: This specific mutation is found in approximately 50% of patients and serves as a critical biomarker for diagnosis and targeted therapy.
The Hallmark Features
To distinguish Langerhans Cell Histiocytosis (Histiocytosis X) from other histiocytic disorders (like Rosai-Dorfman or Hemophagocytic lymphohistiocytosis), we must recognize the specific immunophenotype of the Langerhans Cell Histiocytosis (Histiocytosis X) cell:
- Birbeck Granules: Unique, tennis-racket-shaped cytoplasmic organelles seen under electron microscopy.
- Surface Markers: Pathological cells consistently express CD1a, S100, and Langerin (CD207).
Epidemiology and Clinical Scope
- Incidence: Langerhans Cell Histiocytosis (Histiocytosis X) is primarily a pediatric disease, with a peak incidence between 1 and 3 years of age (approximately 5 - 9 cases per million children). However, it is increasingly recognized in adults, where it often presents with pulmonary involvement related to smoking.
- Spectrum of Disease: Langerhans Cell Histiocytosis (Histiocytosis X) is "the great mimicker" because it can range from a single, self-limiting bone lesion (formerly Eosinophilic Granuloma) to a life-threatening multisystem disease involving "risk organs" like the liver, spleen, and bone marrow.
Pathophysiology: The MAPK Pathway and the "Misguided" Progenitor
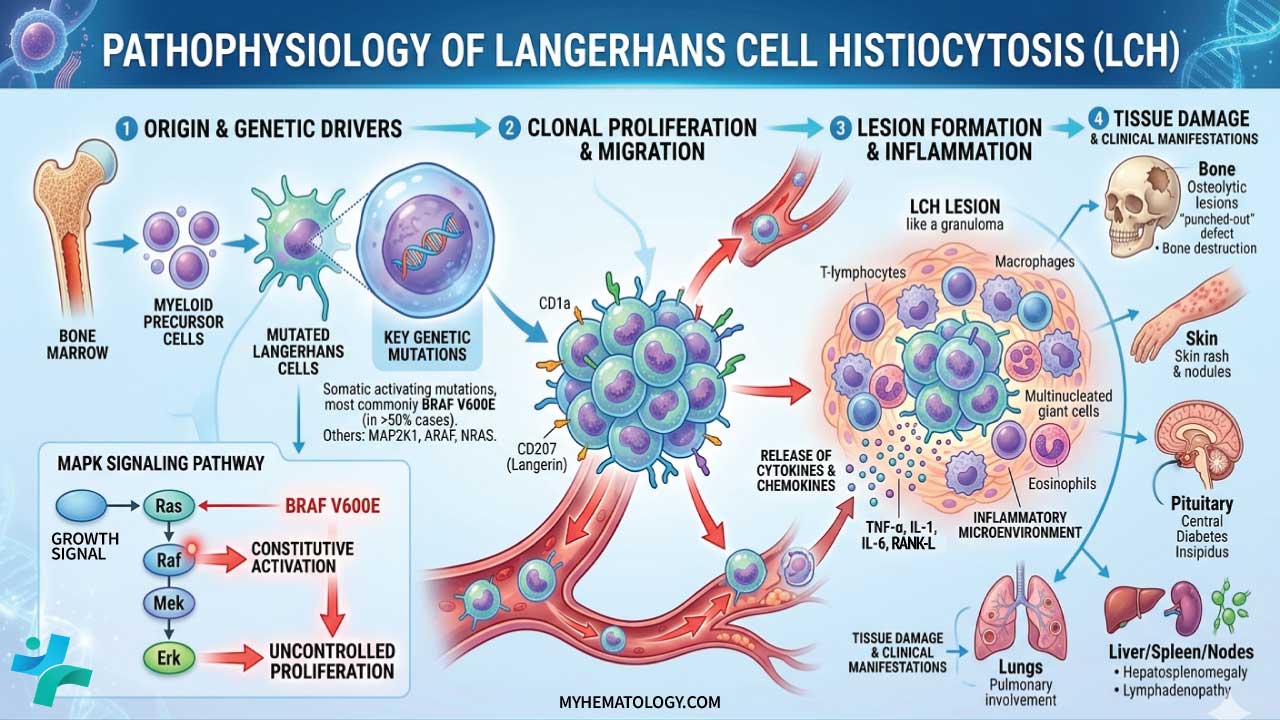
The pathogenesis of Langerhans Cell Histiocytosis (Histiocytosis X) is defined by two main components: somatic mutations in the myeloid lineage and a resulting "cytokine storm" that recruits inflammatory cells.
The "Misguided Myeloid Progenitor" Model
Historically, it was assumed Langerhans Cell Histiocytosis (Histiocytosis X) arose from mature Langerhans cells in the epidermis. We now know Langerhans Cell Histiocytosis (Histiocytosis X) is a systemic myeloid neoplasm.
- The Origin: The disease starts when a hematopoietic progenitor cell in the bone marrow acquires a mutation.
- The "Level" of Mutation: The clinical severity often depends on where in the differentiation process the mutation occurs:
- High-risk/Multisystem LCH: The mutation occurs early in a CD34+ hematopoietic stem cell or a common myeloid progenitor. These mutated cells circulate in the blood and seed multiple organs (liver, spleen, bone marrow).
- Low-risk/Single-system LCH: The mutation occurs later in a more differentiated, tissue-restricted dendritic cell precursor, leading to localized lesions (e.g., a solitary bone lesion).
The MAPK Signaling Pathway
The central "driver" of Langerhans Cell Histiocytosis (Histiocytosis X) is the constitutive activation of the Mitogen-Activated Protein Kinase (MAPK) pathway. This pathway normally regulates cell growth, differentiation, and survival.
- BRAFV600E Mutation: Found in ~50 - 60% of cases. This mutation mimics a permanent "ON" switch for the pathway, leading to uncontrolled cellular proliferation and resistance to apoptosis.
- Other Mutations: In patients without the BRAF mutation, researchers often find mutations in MAP2K1 (which encodes the MEK1 protein) or ARAF and NRAS.
Even without an external growth signal, the pathway remains active, driving the "Langerhans-like" cells to accumulate and persist in tissues where they don't belong.
The "Inflammatory Ringleader" & The Cytokine Storm
Langerhans Cell Histiocytosis (Histiocytosis X) is unique because the "tumor" is not composed entirely of neoplastic cells. In a typical Langerhans Cell Histiocytosis (Histiocytosis X) lesion, the mutated Langerhans Cell Histiocytosis (Histiocytosis X) cells actually make up a minority of the mass (often <10%).
The mutated Langerhans Cell Histiocytosis (Histiocytosis X) cells act as "ringleaders." They secrete high levels of pro-inflammatory cytokines and chemokines, including:
- IL-1, IL-6, and TNF-α
- RANK-L (which activates osteoclasts, leading to the classic "punched-out" lytic bone lesions)
- CCL20 (which recruits more inflammatory cells)
This creates a vicious cycle: the mutated cells recruit normal T-cells, macrophages, and eosinophils. These recruited cells then produce more cytokines that help the mutated Langerhans Cell Histiocytosis (Histiocytosis X) cells survive and proliferate. This "inflammatory milieu" is what causes the actual tissue damage and clinical symptoms.
The Pathophysiologic Spectrum
| Feature | Low-Risk (Single System) | High-Risk (Multisystem) |
| Cell of Origin | Tissue-resident precursor | Early bone marrow progenitor (CD34+) |
| Mutation Site | Downstream/Late | Upstream/Early |
| Typical Organs | Bone, Skin | Liver, Spleen, Bone Marrow |
| Pathway Involved | MAPK/ERK | MAPK/ERK |
Clinical Presentation: The Spectrum of Disease
Bone Involvement (The Most Common Site, ~80%)
Bone lesions are the hallmark of Langerhans Cell Histiocytosis (Histiocytosis X). They are often painful and can occur in any bone, but the skull, femur, ribs, and vertebrae are the most frequent sites.
- The "Punched-Out" Lesion: On imaging, these appear as well-circumscribed lytic lesions without a sclerotic rim.
- Clinical Sign: Localized swelling and exquisite tenderness. In the spine, it can lead to vertebra plana (collapse of the vertebral body), and in the jaw, it may cause "floating teeth" due to alveolar bone destruction.
Skin and Mucous Membranes (~30-40%)
Skin involvement is often the first sign in infants.
- Seborrheic-like Dermatitis: Often mistaken for "cradle cap" or diaper rash, but it is typically more persistent and may have a petechial (hemorrhagic) component.
- Morphology: Scaly papules, nodules, or ulcerations in skin folds (axilla, groin).
- Oral Involvement: Painful gingival swelling or ulcers on the hard palate.
The "Risk Organs" (Critical for Prognosis)
When Langerhans Cell Histiocytosis (Histiocytosis X) involves these three systems, the disease is considered "High Risk," and the 5-year survival rate drops significantly without aggressive systemic therapy:
- Hematopoietic System: Presents as unexplained cytopenias (anemia, thrombocytopenia, or neutropenia) due to bone marrow infiltration or hypersplenism.
- Liver: Hepatomegaly, jaundice, and elevated liver enzymes. A late-stage complication is Sclerosing Cholangitis, which can lead to liver failure.
- Spleen: Splenomegaly, often associated with cytopenias.
Endocrine System
Langerhans Cell Histiocytosis (Histiocytosis X) has a high affinity for the posterior pituitary gland and the hypothalamic-pituitary axis.
- Diabetes Insipidus (DI): The most common endocrine manifestation (polyuria and polydipsia). This is often a permanent deficit even after the Langerhans Cell Histiocytosis (Histiocytosis X) is treated.
- Growth Failure: Due to Growth Hormone deficiency.
Central Nervous System (CNS)
Beyond the pituitary, Langerhans Cell Histiocytosis (Histiocytosis X) can cause a rare but devastating Neurodegenerative Syndrome (LCH-ND).
- Symptoms: Ataxia, dysarthria (slurred speech), tremors, and cognitive decline.
- Timing: This often appears years after the initial diagnosis of systemic Langerhans Cell Histiocytosis (Histiocytosis X).
Pulmonary Langerhans Cell Histiocytosis (Histiocytosis X) (The "Adult" Variant)
While systemic Langerhans Cell Histiocytosis (Histiocytosis X) is usually pediatric, isolated lung involvement is seen primarily in adult smokers.
- Pathophysiology: Smoking-induced inflammation in the small airways recruits mutated myeloid cells.
- Presentation: Dyspnea, non-productive cough, or spontaneous pneumothorax.
- Imaging: Classic "star-shaped" nodules and thin-walled cysts on high-resolution CT.
Clinical Summary Table
| Organ System | Key Clinical Sign | Diagnostic "Clue" |
| Bone | Localized pain/swelling | "Punched-out" lytic lesion on X-ray |
| Skin | Scaly papules/rash | Mimics "cradle cap" but doesn't resolve |
| Pituitary | Polyuria/Polydipsia | Diabetes Insipidus (low urine specific gravity) |
| Liver/Spleen | Jaundice/Abdominal distension | High-risk disease; needs systemic chemo |
| Bone Marrow | Pallor, bruising, infections | Cytopenias (Anemia, Thrombocytopenia) |
Laboratory Investigations and Diagnostic Workup
The diagnosis of Langerhans Cell Histiocytosis (Histiocytosis X) is never made on clinical grounds alone; it requires a combination of laboratory data, imaging, and, most crucially, a tissue biopsy.
Routine Laboratory Baseline
These tests assess the extent of the disease and identify "risk organ" involvement:
- Full Blood Count (FBC) with Differential: To look for cytopenias (anemia, thrombocytopenia, or neutropenia) which suggest bone marrow involvement (high-risk disease).
- Liver Function Tests (LFTs): To screen for hepatomegaly or biliary involvement (hypoalbuminemia, elevated bilirubin, and alkaline phosphatase).
- Coagulation Profile: Specifically PT/APTT and Fibrinogen, as Langerhans Cell Histiocytosis (Histiocytosis X) can occasionally mimic some features of HLH or cause liver-related coagulopathy.
- Urine Osmolality and Water Deprivation Test: Indicated if the patient presents with polyuria/polydipsia to confirm Central Diabetes Insipidus.
The Gold Standard: Tissue Biopsy and Immunohistochemistry (IHC)
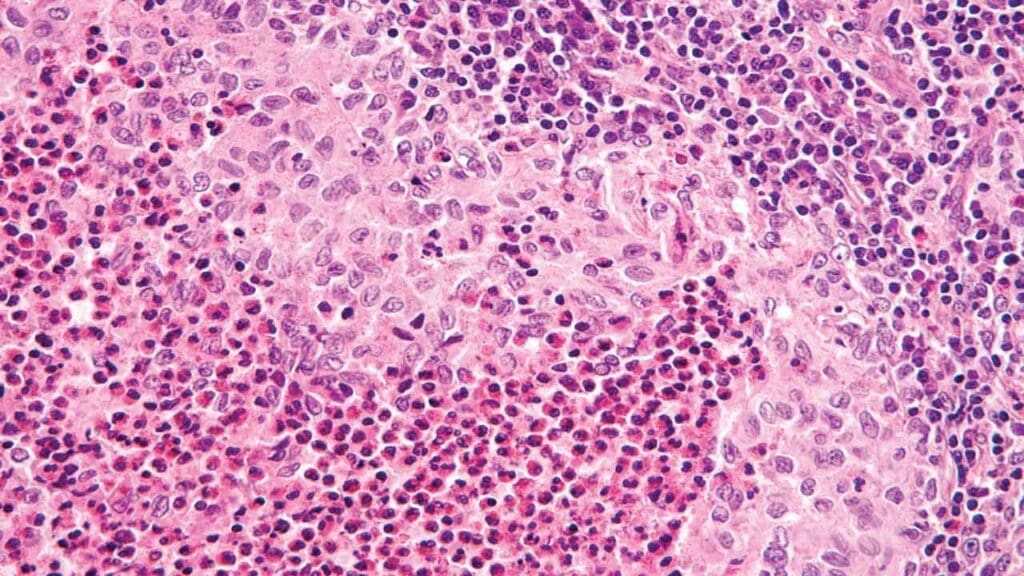
A definitive diagnosis requires histological confirmation from a lesion (usually bone or skin).
- Morphology: Under light microscopy, Langerhans Cell Histiocytosis (Histiocytosis X) cells appear as large, oval cells with "grooved" or "coffee-bean" nuclei, surrounded by a background of eosinophils, lymphocytes, and histiocytes.
- Immunophenotype (The "Must-Know" Markers):
- CD1a and CD207 (Langerin): These are the most specific markers. Positive staining for Langerin is essential as it correlates with the presence of Birbeck granules.
- S100: Almost always positive, though less specific than CD1a/Langerin.
- Electron Microscopy: While rarely used in modern practice due to IHC, identifying Birbeck granules (tennis-racket-shaped organelles) is a pathognomonic finding.
Molecular Testing
With the reclassification of Langerhans Cell Histiocytosis (Histiocytosis X) as a myeloid neoplasm, molecular profiling is now standard:
- BRAFV600E Mutation Testing: Performed on the biopsy tissue. Identifying this mutation not only confirms the clonal nature of the disease but also opens the door for targeted BRAF-inhibitor therapy.
- MAPK Pathway Panel: If BRAF is negative, testing for MAP2K1 mutations is often the next step.
Imaging and Staging
- Skeletal Survey: Conventional X-rays are often superior to bone scans for identifying the characteristic "punched-out" lytic lesions.
- Chest HRCT: Essential in adult smokers to identify the "nodule-to-cyst" progression in pulmonary LCH.
- MRI Brain: Specifically looking for thickening of the pituitary stalk or "bright spot" loss in the posterior pituitary.
Differential Diagnosis
Because Langerhans Cell Histiocytosis (Histiocytosis X) can affect almost any organ, the differential list is broad.
Bone Lesions (The "Lytic Lesion" Diff)
- Osteomyelitis: Often presents with similar pain and swelling, but usually with fever and elevated inflammatory markers (ESR/CRP).
- Ewing Sarcoma: A more aggressive malignant bone tumor; usually has a "moth-eaten" or "onion-skin" periosteal reaction rather than a clean punched-out look.
- Multiple Myeloma: More common in older adults; also presents with lytic lesions.
Skin Lesions (The "Dermatitis" Diff)
- Seborrheic Dermatitis: The most common misdiagnosis in infants ("cradle cap"). Langerhans Cell Histiocytosis (Histiocytosis X) is distinguished by its lack of response to standard topical treatments and the presence of petechiae.
- Atopic Dermatitis / Diaper Rash: Langerhans Cell Histiocytosis (Histiocytosis X) involves the deep skin folds (axilla/groin) and is often more erosive or ulcerative.
Systemic / Multisystem (The "Infiltrative" Diff)
- Hemophagocytic Lymphohistiocytosis (HLH): While both are "histiocytic" disorders, HLH is a medical emergency characterized by high fevers, extreme hyperferritinemia, and cytopenias without the characteristic Langerhans Cell Histiocytosis (Histiocytosis X) tissue lesions.
- Leukemia/Lymphoma: Can present with hepatosplenomegaly and bone marrow failure, but peripheral blood film and marrow flow cytometry will distinguish these.
- Rosai-Dorfman Disease: Another histiocytic disorder, but typically presents with massive, painless lymphadenopathy and is S100+ but CD1a-.
LCH vs. Mimics
| Feature | LCH | Osteomyelitis | Seborrheic Derm. |
| Primary Sign | Punched-out lytic bone lesion | Bone pain + Fever | Scaly scalp/skin rash |
| Key Marker | CD1a+, Langerin+ | Culture positive | Clinical diagnosis |
| Genetics | BRAFV600E | N/A | N/A |
| Response | Chemo/Steroids | Antibiotics | Topical Antifungals |
Disease Classification and Risk Stratification
In modern hematology, Langerhans Cell Histiocytosis (Histiocytosis X) is no longer categorized by historical eponymous syndromes (like Letterer-Siwe). Instead, we use a risk-adapted classification system that dictates treatment intensity and predicts long-term morbidity.
Clinical Classification (The Extent of Disease)
The first step in staging is determining how many systems are involved:
- Single-System LCH (SS-LCH): Involves only one organ or system.
- Unifocal: A single lesion (e.g., one bone lesion).
- Multifocal: Multiple lesions within that same system (e.g., multiple skull lesions).
- Multisystem LCH (MS-LCH): Involves two or more organ systems (e.g., skin and bone).
Risk Stratification (The "Risk Organ" Concept)
The most critical prognostic factor is the involvement of Risk Organs (RO). Patients with MS-LCH are divided into two distinct prognostic groups.
A. High-Risk (RO+) Multisystem LCH
Defined by involvement of one or more of the following:
- Hematopoietic System: Presence of cytopenias (Anemia: Hb < 10 g/dL; Leukopenia: WBC <4.0 × 109/L; Thrombocytopenia: Plt <100 × 109/L).
- Liver: Hepatomegaly (> 3 cm), dysfunction (hypoalbuminemia, hyperbilirubinemia), or sclerosing cholangitis.
- Spleen: Splenomegaly (> 2 cm below costal margin).
Involvement of these organs is associated with a significantly higher mortality rate (historically up to 20%) and requires intensive systemic chemotherapy.
B. Low-Risk (RO-) Multisystem LCH
Involvement of multiple organs (e.g., skin, bone, and lymph nodes) but without liver, spleen, or bone marrow involvement. While these patients have a very low risk of mortality, they have a high risk of disease reactivation (relapse).
Molecular Stratification
As of the latest clinical updates, we are moving away from only looking at "where" the disease is, toward "what" is driving it. This is often referred to as Genotype-Based Stratification.
- BRAF V600E Status: Presence of the BRAFV600E mutation in Peripheral Blood Mononuclear Cells (PBMC) is now recognized as a potent predictor of:
- Treatment Failure: High correlation with poor response to standard vinblastine/prednisone.
- LCH-ND Risk: A 20x higher risk of developing the devastating Neurodegenerative Langerhans Cell Histiocytosis (Histiocytosis X) syndrome.
- The "CNS-Risk" Lesions: Lesions in the craniofacial bones (ear, orbit, temporal bone) are stratified as high-risk for future Central Diabetes Insipidus, even if the patient is currently asymptomatic.
Academic Classification (The "L Group")
The Histiocyte Society (HS) Classification groups Langerhans Cell Histiocytosis (Histiocytosis X) into the "L" (Langerhans) Group.
| Group | Included Disorders |
| L (Langerhans) | LCH, Indeterminate Cell Histiocytosis, Erdheim-Chester Disease (some variants). |
| C (Cutaneous) | Xanthogranuloma family. |
| R (RNA-related) | Rosai-Dorfman Disease. |
| M (Malignant) | Histiocytic Sarcoma. |
| H (Hemophagocytic) | Primary and Secondary HLH. |
Treatment and Management Strategies
The management of Langerhans Cell Histiocytosis (Histiocytosis X) is a perfect example of risk-adapted therapy. Because the disease can range from a single "punched-out" bone lesion to a fatal systemic failure, the intensity of the treatment must match the "risk" level of the patient.
The primary goal of therapy is twofold: to save lives in high-risk patients and to prevent reactivations (relapses) and permanent sequelae (like Diabetes Insipidus) in low-risk patients.
Low-Risk: Single-System LCH (SS-LCH)
For many patients with isolated involvement, "less is more."
- Observation: In some cases, such as "congenital self-healing reticulohistiocytosis" (skin-only LCH in neonates), the lesions may regress spontaneously under close monitoring.
- Local Therapy:
- Bone: A single lytic lesion is often treated with curettage (surgical scraping) or a local injection of methylprednisolone.
- Skin: Isolated skin LCH may respond to topical steroids or nitrogen mustard.
- When to use Systemic Therapy: If the lesion is in a "CNS-risk" location (e.g., the skull base or orbit) or if there are multiple bone lesions, systemic treatment is started to prevent the development of Diabetes Insipidus.
The Standard: Vinblastine and Prednisone (LCH-IV Protocol)
For all multisystem cases and multifocal single-system cases, the Vinblastine + Prednisone regimen remains the global standard of care.
- The Initial Phase (6–12 weeks):
- Vinblastine: 6 mg/m2 IV bolus weekly.
- Prednisone: 40 mg/m2/day orally, usually tapered after the first 4 weeks.
- The Continuation Phase: If the patient responds well, they move to a maintenance phase (pulses every 3 weeks) for a total treatment duration of 12 months.
- Recent trials (LCH-IV) show that extending treatment to 12 months (instead of 6) significantly reduces the high rate of disease reactivation.
Targeted Therapy
The discovery of the MAPK pathway mutations (like BRAF V600E) has revolutionized the management of refractory (resistant) Langerhans Cell Histiocytosis (Histiocytosis X).
- BRAF Inhibitors (e.g., Vemurafenib, Dabrafenib): These oral drugs "switch off" the mutated protein. They are highly effective, often showing dramatic clinical improvement within days in high-risk infants who have failed chemotherapy.
- MEK Inhibitors (e.g., Trametinib, Cobimetinib): These act further downstream in the pathway. They are particularly useful for patients who are BRAF-negative but have other MAPK mutations (like MAP2K1).
While targeted therapies are powerful, the disease often recurs once the drug is stopped. Research is currently focused on how to combine these with traditional chemo to achieve a permanent cure.
Salvage Therapy for High-Risk Non-Responders
If a patient has "Risk Organ" involvement (liver, spleen, or bone marrow) and does not respond to the first 6 weeks of standard chemo, they are at high risk of mortality.
- Nucleoside Analogs: Drugs like Cladribine (2-CdA) or Clofarabine are used as potent second-line agents.
- Hematopoietic Stem Cell Transplant (HSCT): Reserved for the most severe, refractory cases as a final attempt to replace the mutated myeloid progenitor clones.
Treatment Overview
| Disease Category | Primary Treatment | Duration | Goal |
| SS-LCH (Isolated Bone) | Curettage or Local Steroids | N/A | Local Control |
| MS-LCH (Low-Risk) | Vinblastine + Prednisone | 12 Months | Prevent Reactivation |
| MS-LCH (High-Risk) | Intensified Chemo + MAPK Inhibitors | 12+ Months | Survival / Remission |
| Refractory LCH | Targeted Therapy / Nucleoside Analogs | Indefinite/Bridge to HSCT | Disease Control |
Long-Term Follow-Up Strategy
Surveillance must be multidisciplinary. Even after complete clinical response (CCR), patients require "indefinite" monitoring for late effects.
Surveillance Schedule
- Years 1–2 (Post-Treatment): Clinical evaluation and laboratory tests (CBC, LFTs) every 3 months.
- Years 3–5: Visits every 6 months.
- Beyond 5 Years: Annual visits, often transitioning to a specialized adult survivorship clinic.
Key Monitoring Parameters
- Endocrine: Annual screening for diabetes insipidus (morning urine osmolality) and growth velocity. If the pituitary stalk was thickened at diagnosis, monitor anterior pituitary hormones (GH, TSH, Cortisol).
- Neurological: Annual neurological exam focusing on ataxia or tremors. For "CNS-risk" patients, consider a baseline Brain MRI 1 year post-treatment to screen for pre-symptomatic ND-LCH.
- Auditory: Periodic audiometry for those with history of ear/mastoid involvement.
- Imaging:
- PET/CT or Skeletal Survey: Only if new symptoms (bone pain, rash) arise. Routine "surveillance" imaging of bones is generally avoided to limit radiation.
- Chest CT: Annual or biennial for smokers or those with previous pulmonary LCH to monitor for cystic progression or secondary malignancies.
Frequently Asked Questions (FAQs)
Is Langerhans Cell Histiocytosis a type of cancer?
Yes, it is now officially classified by the WHO as an inflammatory myeloid neoplasm. While it behaves differently than a typical "liquid" or "solid" tumor, its clonal nature and genetic mutations (like BRAFV600E) align it with neoplastic disorders.
Why was it previously called "Histiocytosis X"?
The "X" stood for the unknown origin of the proliferating cells. In 1953, it was recognized as a single disease entity, but it wasn't until the discovery of Birbeck granules that the cell was identified as a Langerhans-like cell, leading to the name change in the 1980s.
Does Langerhans Cell Histiocytosis (Histiocytosis X) only affect children?
While it is most common in children aged 1–3, it can affect patients of any age. Adult Langerhans Cell Histiocytosis (Histiocytosis X) often presents uniquely, frequently as Pulmonary LCH in smokers.
Can Langerhans Cell Histiocytosis (Histiocytosis X) be cured with surgery alone?
Only in very specific cases. A single, isolated bone lesion may be treated with surgical curettage or local steroid injection. However, most multifocal or multisystem cases require systemic chemotherapy to prevent recurrence.
What is the most common permanent complication?
Central Diabetes Insipidus (DI) is the most frequent permanent sequela, occurring in up to 30% of patients due to infiltration of the posterior pituitary or hypothalamic axis.
Glossary of Related Medical Terms
- Birbeck Granules: Unique, rod-shaped cytoplasmic organelles with a striated center and a "tennis racket" appearance, found exclusively in Langerhans cells.
- CD1a / CD207 (Langerin): Specific protein markers expressed on the surface of Langerhans cells, used in immunohistochemistry to confirm an LCH diagnosis.
- Cytokine Storm: A massive release of pro-inflammatory proteins (IL-1, IL-6, TNF-α) by LCH cells that recruits other immune cells and causes tissue damage.
- Lytic Lesion: An area of bone destruction that appears as a "dark hole" or "punched-out" area on an X-ray due to localized osteoclast activation.
- MAPK/ERK Pathway: A major signaling pathway that transmits signals from the cell surface to the nucleus. Mutations in this pathway cause the "always-on" growth signal in LCH.
- Neurodegenerative LCH (ND-LCH): A rare, late-onset complication involving the progressive loss of neurons in the cerebellum and basal ganglia, leading to ataxia and cognitive changes.
- Risk Organs: A clinical term for the liver, spleen, and bone marrow; involvement of these organs significantly increases the risk of mortality.
- Vertebra Plana: A radiographic finding where a vertebral body collapses into a thin, flat disk, often caused by an LCH eosinophilic granuloma in the spine.
Disclaimer: This article is intended for informational purposes only and is specifically targeted towards medical students. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Tillotson CV, Reynolds SB, Patel BC. Langerhans Cell Histiocytosis. [Updated 2024 Apr 18]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2026 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK430885/
- Rodriguez-Galindo, C., & Allen, C. E. (2020). Langerhans cell histiocytosis. Blood, 135(16), 1319–1331. https://doi.org/10.1182/blood.2019000934
- Sconocchia, T., Foßelteder, J., Sconocchia, G., & Reinisch, A. (2023). Langerhans cell histiocytosis: current advances in molecular pathogenesis. Frontiers in immunology, 14, 1275085. https://doi.org/10.3389/fimmu.2023.1275085
- McKinney, R. A., & Wang, G. (2025). Langerhans Cell Histiocytosis and Other Histiocytic Lesions. Head and neck pathology, 19(1), 26. https://doi.org/10.1007/s12105-025-01766-2
- Radzikowska E. (2017). Pulmonary Langerhans' cell histiocytosis in adults. Advances in respiratory medicine, 85(5), 277–289. https://doi.org/10.5603/ARM.a2017.0046
- Goyal, G., Tazi, A., Go, R. S., Rech, K. L., Picarsic, J. L., Vassallo, R., Young, J. R., Cox, C. W., Van Laar, J., Hermiston, M. L., Cao, X. X., Makras, P., Kaltsas, G., Haroche, J., Collin, M., McClain, K. L., Diamond, E. L., & Girschikofsky, M. (2022). International expert consensus recommendations for the diagnosis and treatment of Langerhans cell histiocytosis in adults. Blood, 139(17), 2601–2621. https://doi.org/10.1182/blood.2021014343
- Khoury, J.D., Solary, E., Abla, O. et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 36, 1703–1719 (2022). https://doi.org/10.1038/s41375-022-01613-1
- Emile, J. F., Abla, O., Fraitag, S., Horne, A., Haroche, J., Donadieu, J., Requena-Caballero, L., Jordan, M. B., Abdel-Wahab, O., Allen, C. E., Charlotte, F., Diamond, E. L., Egeler, R. M., Fischer, A., Herrera, J. G., Henter, J. I., Janku, F., Merad, M., Picarsic, J., Rodriguez-Galindo, C., … Histiocyte Society (2016). Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood, 127(22), 2672–2681. https://doi.org/10.1182/blood-2016-01-690636