TL;DR
Wilson’s disease is a rare genetic disorder causing toxic copper accumulation in the liver, brain, and other organs.
- Pathophysiology ▾: Defective ATP7B gene impairs copper excretion into bile and incorporation into ceruloplasmin, leading to copper overload and cellular damage.
- Symptoms ▾: Highly variable; can include liver disease (jaundice, fatigue), neurological issues (tremors, speech problems), psychiatric changes, and Kayser-Fleischer rings in the eyes.
- Laboratory Investigation ▾: Low serum ceruloplasmin, high urinary copper, elevated hepatic copper (biopsy), genetic testing (ATP7B mutations), and Kayser-Fleischer ring detection via slit lamp.
- Treatment & Management ▾: Lifelong chelation therapy (penicillamine, trientine), zinc salts, low-copper diet, symptomatic management, and liver transplantation in severe cases.
- Potential Complications ▾: Cirrhosis, liver failure, neurological damage, psychiatric disorders, kidney problems, hemolytic anemia, skeletal issues, and can be fatal if untreated.
*Click ▾ for more information
Introduction
Wilson’s disease is a rare, inherited disorder characterized by the abnormal accumulation of copper in the body, primarily in the liver, brain, and cornea. This buildup of excess copper leads to cellular damage and dysfunction in these organs, resulting in a wide range of clinical manifestations, predominantly affecting the liver (causing liver disease) and the nervous system (causing neurological and psychiatric symptoms).
The underlying cause is a mutation in the ATP7B gene, which is responsible for producing a protein that plays a crucial role in transporting copper out of liver cells and incorporating it into ceruloplasmin, the main copper-carrying protein in the bloodstream. Due to the faulty protein, the liver’s ability to excrete excess copper into bile is impaired, and less copper is bound to ceruloplasmin, leading to its toxic accumulation in various tissues.
While Wilson’s disease is primarily known for its hepatic and neurological effects, it has significant associations with hematology. The excess copper in Wilson’s disease can directly and indirectly impact the components of blood and the bone marrow, leading to several hematological abnormalities.
Copper’s Role in Hematopoiesis and the Impact of Copper Overload
Normal Copper Metabolism
Copper is an essential trace element, meaning the body needs it in small amounts to function properly. The average adult body contains about 50-150 mg of copper.
Dietary copper, primarily from sources like shellfish, nuts, seeds, legumes, and organ meats, is absorbed mainly in the stomach and upper small intestine (duodenum). Copper is ingested in the Cu2+ (cupric) form but needs to be reduced to Cu1+ (cuprous) for absorption. This reduction is facilitated by enzymes like duodenal cytochrome b reductase (DCYTB) and possibly aided by vitamin C.
In the bloodstream, copper binds to several proteins for transport to the liver and other tissues. The primary transporter is ceruloplasmin, a protein synthesized in the liver. Ceruloplasmin binds about 70-95% of the copper in plasma. Albumin and transcuprein (also known as alpha-2-macroglobulin) bind the remaining copper, mainly during the initial transport from the gut to the liver.
The liver is the central organ in copper metabolism. Hepatocytes take up copper from the blood via CTR1 and other transporters. Inside the liver cells, copper is chaperoned by specific proteins that direct it to various pathways:
- Incorporation into Ceruloplasmin: Copper is incorporated into apoceruloplasmin to form functional ceruloplasmin, which is then secreted back into the bloodstream. This process is facilitated by the ATP7B protein.
- Storage: Copper can be stored in hepatocytes bound to metallothionein.
- Incorporation into Cuproenzymes: Copper is essential as a cofactor for numerous enzymes involved in various metabolic processes, including energy production (cytochrome c oxidase), antioxidant defense (superoxide dismutase), neurotransmitter synthesis (dopamine-beta-hydroxylase), and connective tissue formation (lysyl oxidase).
- Biliary Excretion: The liver is the primary route for eliminating excess copper from the body. ATP7B plays a crucial role in transporting copper into the bile for excretion in feces.
- Distribution to Other Tissues: Ceruloplasmin in the blood delivers copper to other tissues throughout the body, where it is taken up by cells via CTR1 and other mechanisms to fulfill their specific metabolic needs.
Function of Copper in Normal Hematopoiesis
Hematopoiesis is the process of blood cell formation, including red blood cells (erythrocytes), white blood cells (leukocytes), and platelets (thrombocytes). It primarily occurs in the bone marrow. Copper plays a vital, though indirect, role in this process:
- Iron Metabolism: Copper is essential for proper iron metabolism, which is critical for erythropoiesis (red blood cell production). Ceruloplasmin has ferroxidase activity, meaning it helps oxidize ferrous iron (Fe2+) to ferric iron (Fe3+), the form that can bind to transferrin, the main iron transport protein in the blood. This oxidation is necessary for iron to be loaded onto transferrin for delivery to the bone marrow, where it is incorporated into hemoglobin in developing red blood cells. Another copper-containing ferroxidase, hephaestin, is crucial for iron export from enterocytes into the bloodstream.
- Enzyme Cofactor: Copper is a cofactor for enzymes like superoxide dismutase, which may play a role in protecting hematopoietic cells from oxidative damage during their development. Cytochrome c oxidase, another copper-dependent enzyme, is essential for cellular respiration, providing energy for the rapidly dividing hematopoietic cells.
In essence, while copper is not a direct building block of blood cells like iron, it is crucial for the enzymes and processes that support their production, particularly iron handling and cellular respiration within the bone marrow. Disruptions in copper metabolism, as seen in Wilson’s disease, can therefore have significant consequences for hematopoiesis.
Pathophysiology of Wilson’s Disease
The pathophysiology of Wilson’s disease centers around a genetic defect in copper metabolism, leading to the toxic accumulation of copper primarily in the liver, brain, and cornea.
Wilson’s disease is an autosomal recessive disorder caused by mutations in the ATP7B gene located on chromosome 13. This gene encodes for a P-type ATPase copper transporter protein primarily found in hepatocytes (liver cells) and also in other tissues like the brain.
The ATP7B protein has two crucial functions in the liver:
- Biliary Excretion of Copper: It facilitates the transport of excess copper into the bile for elimination from the body via feces. In Wilson’s disease, this function is impaired, leading to reduced copper excretion.
- Incorporation of Copper into Ceruloplasmin: It is necessary for the incorporation of copper into apoceruloplasmin (an inactive protein) to form functional ceruloplasmin, the main copper-carrying protein in the bloodstream. In Wilson’s disease, this process is also defective.
Due to the impaired biliary excretion, copper begins to accumulate within the hepatocytes from birth. The reduced formation of ceruloplasmin leads to lower levels of circulating ceruloplasmin. While the total serum copper might appear low or normal due to decreased ceruloplasmin-bound copper, the amount of free, unbound copper in the blood increases.
As the liver’s capacity to store copper is eventually exceeded, the toxic free copper spills over into the bloodstream and is deposited in other organs.
Organ Damage Due to Copper Toxicity
- Liver: The initial site of copper accumulation. This can lead to a spectrum of liver disease, ranging from asymptomatic elevation of liver enzymes and fatty liver to chronic hepatitis, fibrosis, cirrhosis, and even fulminant hepatic failure. The mechanisms of liver damage involve oxidative stress, mitochondrial dysfunction, and direct cellular injury caused by the excess copper.
- Brain: Copper deposits primarily in the basal ganglia (especially the lenticular nucleus: putamen and globus pallidus), thalamus, and brainstem. This can disrupt normal neurological function, leading to a variety of movement disorders (tremors, dystonia, dysarthria, ataxia), psychiatric symptoms (personality changes, depression, anxiety, psychosis), and cognitive impairment. The exact mechanisms of neuronal damage are complex and involve oxidative stress, excitotoxicity, and impaired neurotransmitter metabolism.
- Cornea: Copper deposition in Descemet’s membrane of the cornea results in the characteristic Kayser-Fleischer rings, which are a crucial diagnostic sign.
- Other Organs: While less prominent, copper can also accumulate in other tissues like the kidneys (leading to renal tubular dysfunction), heart (rarely causing cardiomyopathy), and joints (arthropathy).
Associated Hematological Problem
While Wilson’s disease is primarily known for its hepatic and neurological effects, it has significant associations with hematology, the branch of medicine concerned with the study of blood and blood disorders. The excess copper in Wilson’s disease can directly and indirectly impact the components of blood and the bone marrow, leading to several hematological abnormalities.
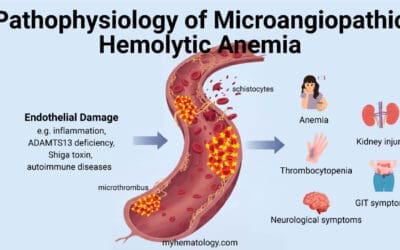
- Hemolytic Anemia: One of the most recognized hematological complications is hemolytic anemia, a condition where red blood cells are prematurely destroyed. The high levels of free (unbound) copper in the bloodstream can directly damage the membranes of red blood cells, making them fragile and prone to lysis (rupture). This can manifest as sudden episodes of acute hemolytic anemia, sometimes coinciding with acute liver decompensation.
- Anemia of Chronic Disease: The chronic inflammation associated with liver disease in Wilson’s can also contribute to anemia of chronic disease, a type of anemia characterized by impaired iron utilization.
- Leukopenia and Thrombocytopenia: In some instances, excess copper can potentially lead to leukopenia (low white blood cell count) and thrombocytopenia (low platelet count). This may be due to direct toxic effects on the bone marrow or indirectly due to hypersplenism (enlargement of the spleen) in patients with advanced liver disease, leading to increased sequestration and destruction of blood cells. However, these findings are less consistent than hemolytic anemia.
- Coagulation Abnormalities: Liver dysfunction, a common feature of Wilson’s disease, can impair the synthesis of coagulation factors, potentially leading to abnormalities in blood clotting.
Signs and Symptoms of Wilson’s Disease
The signs and symptoms of Wilson’s disease are highly variable and depend on which organs are primarily affected by copper accumulation. They can also vary with the age of onset. It’s important to note that symptoms may be subtle initially and can mimic those of other conditions, leading to diagnostic challenges.
Hepatic Manifestations (Liver-related)
These are often the initial symptoms, particularly in children and young adults.
- Asymptomatic Liver Disease: Many individuals may initially have only mild, non-specific symptoms or even no noticeable symptoms, with the disease being detected through abnormal liver function tests.
- Fatigue and Weakness
- Loss of Appetite
- Nausea and Vomiting
- Abdominal Pain or Swelling: Discomfort or distension in the upper abdomen due to liver enlargement or fluid buildup (ascites in advanced stages).
- Jaundice: Yellowing of the skin and whites of the eyes due to elevated bilirubin levels, indicating liver dysfunction.
- Easy Bruising or Bleeding: Impaired liver function can affect the production of clotting factors.
- Spider Angiomas: Small, spider-like blood vessels visible on the skin.
- Hepatomegaly and Splenomegaly
- Acute Hepatitis: Sudden onset of liver inflammation, which can range from mild to severe and even lead to fulminant hepatic failure.
- Cirrhosis: Chronic scarring of the liver, leading to impaired liver function and potential complications like portal hypertension, esophageal varices (swollen blood vessels in the esophagus that can bleed), and ascites.
Neurological Manifestations (Brain-related)
These are more common in individuals diagnosed in later adolescence or adulthood.
- Movement Disorders:
- Tremors: Shaking, often of the hands or arms.
- Dystonia: Sustained muscle contractions causing twisting and repetitive movements or abnormal postures.
- Dysarthria: Difficulty with speech due to problems with the muscles used for speaking (slurred or slow speech).
- Dysphagia: Difficulty swallowing.
- Ataxia: Lack of coordination and balance, leading to clumsy movements and gait abnormalities.
- Chorea: Involuntary, jerky movements.
- Rigidity: Stiffness of muscles.
- Cognitive Impairment: Difficulty with memory, concentration, and executive functions.
- Behavioral and Psychiatric Changes:
- Personality Changes: Irritability, impulsivity, social withdrawal.
- Depression and Anxiety: Persistent low mood and excessive worry.
- Psychosis: In severe cases, hallucinations or delusions.
- Emotional Lability: Rapid and exaggerated changes in mood.
Kayser-Fleischer Rings (Eye-related)
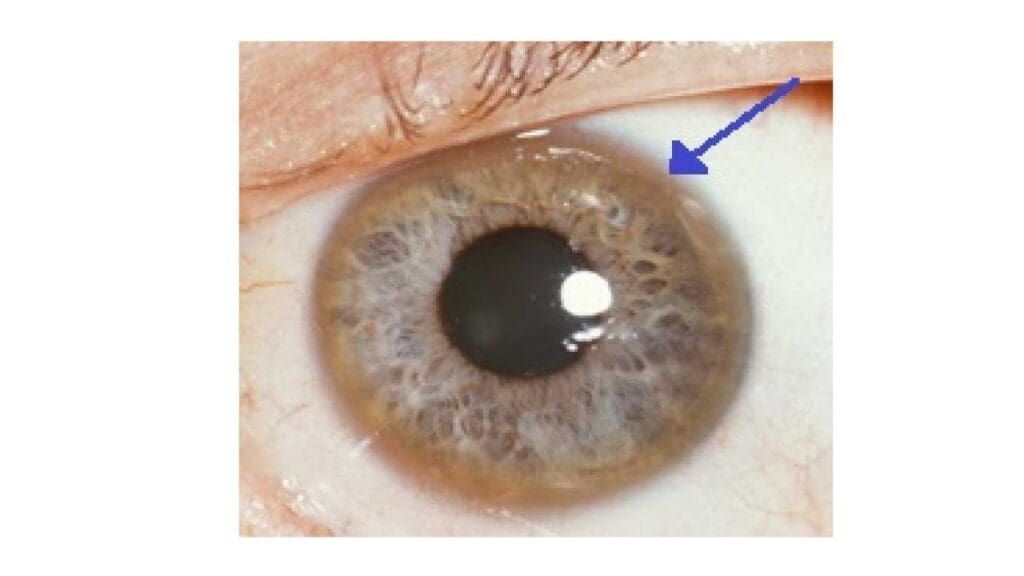
These are characteristic golden-brown or greenish rings around the iris (the colored part) of the eyes. They are caused by copper deposits in Descemet’s membrane of the cornea. They are a crucial diagnostic sign but require a slit-lamp examination by an ophthalmologist to be visualized. While highly suggestive of Wilson’s disease, they may be absent in some individuals, particularly those with only liver involvement or early-stage disease.
Other Potential Signs and Symptoms
- Hematological Abnormalities: As discussed previously, including hemolytic anemia, and less commonly, leukopenia or thrombocytopenia.
- Renal Manifestations: Kidney problems such as renal tubular acidosis, kidney stones, and aminoaciduria.
- Cardiac Manifestations: Rarely, cardiomyopathy or arrhythmias.
- Skeletal Manifestations: Joint pain (arthralgia), arthritis, and osteoporosis.
- Endocrine Abnormalities: Menstrual irregularities in women, and rarely, hypoparathyroidism.
- “Sunflower Cataracts”: Less common than Kayser-Fleischer rings, these are distinctive multicolored cataracts with a spoke-like pattern.
- Lunulae Ceruleae: Bluish discoloration at the base of the fingernails (less common and not specific to Wilson’s disease).
The variability in presentation underscores the importance of considering Wilson’s disease in the differential diagnosis of a wide range of hepatic, neurological, and psychiatric conditions, especially in younger individuals. A combination of clinical findings, laboratory tests, and ophthalmological examination is usually required for accurate diagnosis.
Laboratory Investigations of Wilson’s Disease
The laboratory investigations for Wilson’s disease are crucial for diagnosis and monitoring treatment. No single test is definitive, and the diagnosis often relies on a combination of findings.
Copper Metabolism Studies
- Serum Ceruloplasmin: Typically, levels are low (below 20 mg/dL; normal range is usually 20-40 mg/dL). This reflects the impaired incorporation of copper into apoceruloplasmin due to the defective ATP7B protein. Low ceruloplasmin can also be seen in other conditions like malnutrition, malabsorption, nephrotic syndrome, and some other liver diseases. Importantly, about 5-10% of Wilson’s disease patients, especially those with liver involvement only, may have normal or near-normal ceruloplasmin levels, particularly if they present with acute hepatitis (as ceruloplasmin is an acute phase reactant and can be transiently elevated).
- Serum Copper (Total): Levels are often low because most copper in the blood is normally bound to ceruloplasmin. However, this test alone is not very sensitive or specific. In acute liver failure due to Wilson’s disease, total serum copper can paradoxically be high due to the release of large amounts of copper from the damaged liver.
- Serum Free Copper (Non-Ceruloplasmin Bound Copper): This level is typically elevated in Wilson’s disease because less copper is being incorporated into ceruloplasmin, leading to a higher concentration of toxic free copper circulating in the blood. This is often a more helpful marker than total serum copper.
- 24-Hour Urinary Copper Excretion: Usually markedly elevated (typically >100 mcg/24 hours; normal is <40 mcg/24 hours). This reflects the increased amount of free copper in the blood being filtered by the kidneys. In some cases where the diagnosis is uncertain, a 24-hour urine copper collection may be performed after administering a chelating agent like D-penicillamine. A significant increase in urinary copper excretion after penicillamine administration supports the diagnosis of Wilson’s disease.
- Hepatic Copper Concentration (Liver Biopsy): This is considered the gold standard for diagnosing Wilson’s disease. A copper concentration greater than 250 micrograms per gram of dry liver weight is highly suggestive of Wilson’s disease. Copper distribution in the liver can be uneven, so in rare cases, a biopsy might miss areas of high copper concentration. However, it provides valuable information about the degree of copper accumulation and the extent of liver damage (histology).
Liver Function Tests (LFTs)
LFTs are often abnormal, reflecting liver damage. Patterns can vary from mild elevations to those seen in acute hepatitis or chronic liver disease. Alkaline phosphatase levels may be disproportionately low in Wilson’s disease-related acute liver failure. Albumin levels may be low, and prothrombin time prolonged in advanced liver disease due to impaired synthetic function.
Hematological Tests
- Complete Blood Count (CBC) and Peripheral Blood Smear: To assess for anemia (low red blood cell count), particularly hemolytic anemia (presence of schistocytes or other signs of red blood cell fragmentation).
- Reticulocyte Count: Elevated in hemolytic anemia, indicating increased red blood cell production by the bone marrow.
- Hemolysis Markers: Elevated indirect bilirubin, LDH, and decreased haptoglobin can support the presence of hemolysis.
- Coombs Test (Direct Antiglobulin Test): Typically negative in Wilson’s disease-related hemolytic anemia, as it is usually non-immune mediated.
Ophthalmological Examination
- Slit-Lamp Examination: Essential for detecting Kayser-Fleischer rings, the characteristic copper deposits in the cornea. Their presence, especially in individuals with neurological or hepatic symptoms, strongly supports the diagnosis.
Neurological Investigations (if neurological symptoms are present)
- Magnetic Resonance Imaging (MRI) of the Brain: May show characteristic abnormalities, particularly T2-weighted hyperintensities in the basal ganglia (especially the lenticular nucleus) and brainstem. While not specific to Wilson’s disease, these findings in the context of other clinical and laboratory features are suggestive. The “face of the giant panda” sign in the midbrain can also be seen in some cases.
Genetic Testing
- ATP7B Gene Mutation Analysis: Identifying two pathogenic mutations in the ATP7B gene confirms the diagnosis of Wilson’s disease.It is also crucial for screening asymptomatic family members of affected individuals to identify those who may be presymptomatic or carriers.
Diagnostic Approach
The diagnosis of Wilson’s disease often involves a combination of these tests, along with a thorough clinical evaluation and family history. The Leipzig criteria are often used to aid in the diagnosis, assigning points based on clinical signs (Kayser-Fleischer rings, neurological symptoms, hepatic symptoms), biochemical markers (low ceruloplasmin, elevated urinary copper, elevated hepatic copper), and genetic testing.
Treatment and Management of Wilson’s Disease
The treatment and management of Wilson’s disease aim to reduce the accumulated copper in the body, prevent further copper buildup, and manage the associated symptoms. This typically involves a lifelong commitment to therapy and regular monitoring.
Goals of Treatment
- Reduce Copper Overload: The primary goal is to remove the excess copper that has accumulated in the liver, brain, and other organs.
- Prevent Re-accumulation: Once copper levels are reduced, ongoing treatment is necessary to prevent it from building up again.
- Manage Symptoms: Address the specific hepatic, neurological, and psychiatric manifestations of the disease.
- Prevent Complications: Minimize the risk of long-term organ damage and improve the patient’s quality of life.
Pharmacological Therapy (Chelating Agents)
These medications bind to copper in the body, allowing it to be excreted in the urine.
- D-Penicillamine: A potent chelating agent that promotes urinary excretion of copper. It can have significant side effects, including hypersensitivity reactions (rash, fever), gastrointestinal upset, proteinuria, blood dyscrasias (anemia, leukopenia, thrombocytopenia), and neurological worsening (paradoxical reaction, especially initially). It requires close monitoring for side effects with regular blood tests and urine analysis. Pyridoxine (vitamin B6) supplementation is usually recommended as D-penicillamine can interfere with vitamin B6 metabolism.
- Trientine (Triethylene Tetramine): Another chelating agent that binds copper and promotes its urinary excretion. Generally better tolerated than D-penicillamine, with fewer and less severe side effects, mainly mild gastrointestinal upset.
- Tetrathiomolybdate: Works differently by blocking copper absorption in the gut and binding copper in the blood, making it unavailable for tissue uptake and promoting its excretion in the stool. It is often used as initial therapy in patients with neurological symptoms as it may lead to a faster improvement compared to penicillamine or trientine. It can also be used as a bridge to liver transplantation in severely ill patients.
Pharmacological Therapy (Zinc Salts)
Zinc interferes with copper absorption in the gastrointestinal tract and also induces the production of metallothionein in intestinal cells, which binds copper and prevents its absorption. It is primarily used for maintenance therapy after decoppering has been achieved with chelating agents and in presymptomatic individuals identified through family screening. It can also be used as primary therapy in patients with mild or no symptoms.
Dietary Management
While medication is the cornerstone of treatment, dietary modifications can help reduce copper intake.
Liver Transplantation
Liver transplantation is a life-saving option for patients with:
- Fulminant Hepatic Failure: Acute liver failure due to Wilson’s disease.
- Decompensated Cirrhosis: Advanced liver disease with severe complications unresponsive to medical therapy.
- Severe Neurological Disease: In some cases where neurological symptoms are debilitating and not improving with medical treatment.
Liver transplantation replaces the diseased liver with a healthy one, effectively correcting the underlying metabolic defect. Long-term immunosuppression is required after transplantation.
Symptomatic Management
- Neurological Symptoms: Medications (e.g., for tremor, dystonia), physical therapy, occupational therapy, and speech therapy can help manage neurological manifestations.
- Psychiatric Symptoms: Depression, anxiety, or psychosis should be treated with appropriate medications and psychological support.
- Liver-related Complications: Manage ascites, variceal bleeding, and other complications of cirrhosis as needed.
Long-Term Management and Monitoring
Wilson’s disease requires lifelong treatment and regular monitoring.
Potential Complications of Wilson’s Disease
If left untreated or poorly managed, Wilson’s disease can lead to a range of serious and potentially life-threatening complications due to the progressive accumulation of copper in various organs.
Liver-Related Complications
- Cirrhosis: Chronic copper accumulation leads to inflammation and scarring (fibrosis) of the liver tissue. Over time, this can progress to cirrhosis, severely impairing liver function.
- Liver Failure: The progressive damage from cirrhosis can eventually lead to liver failure, where the liver can no longer perform its vital functions. This can occur acutely or develop gradually over years.
- Acute Liver Failure: In some cases, Wilson’s disease can present with a sudden and severe form of liver failure, sometimes accompanied by hemolytic anemia. This is a medical emergency.
- Hepatic Encephalopathy: As the liver fails to remove toxins from the blood, these toxins can build up and affect brain function, leading to confusion, drowsiness, and even coma.
- Increased Risk of Liver Cancer (Hepatocellular Carcinoma): While the risk is lower than in other causes of cirrhosis, it is still elevated in individuals with Wilson’s disease-related cirrhosis.
- Portal Hypertension and its complications: Scarring of the liver can impede blood flow, leading to increased pressure in the portal vein. This can cause complications like esophageal varices (swollen blood vessels in the esophagus that can rupture and bleed), ascites (fluid buildup in the abdomen), and splenomegaly (enlarged spleen).
Neurological and Psychiatric Complications
- Permanent Neurological Damage: While treatment can often improve neurological symptoms, some individuals may experience lasting neurological deficits, such as persistent tremors, dystonia, or coordination problems, especially if treatment is delayed.
- Severe Disability: Untreated neurological involvement can lead to severe physical disability, making it difficult to perform daily activities.
- Progressive Dementia: Cognitive impairment can worsen over time if copper continues to accumulate in the brain, potentially leading to dementia in severe, untreated cases.
- Severe Psychiatric Disorders: Untreated Wilson’s disease can result in significant and debilitating psychiatric issues, including severe depression, psychosis, and personality changes.
- Suicidal Ideation: The psychological impact of the disease and copper accumulation in the brain can increase the risk of suicidal thoughts and behavior.
Other Complications
- Kidney Problems: Copper accumulation can damage the kidneys, leading to issues like renal tubular acidosis, kidney stones, and aminoaciduria (excessive excretion of amino acids in the urine).
- Hemolytic Anemia: As discussed earlier, high levels of free copper can cause the premature destruction of red blood cells, leading to anemia and jaundice.
- Skeletal Problems: Wilson’s disease can be associated with joint pain (arthralgia), arthritis, and weakened bones (osteoporosis or osteopenia), increasing the risk of fractures.
- Cardiac Problems: In rare cases, copper accumulation can affect the heart, leading to cardiomyopathy (weakening of the heart muscle) or arrhythmias.
- Menstrual Irregularities and Infertility: In women, liver dysfunction and hormonal imbalances due to Wilson’s disease can cause menstrual problems and difficulties with fertility, as well as an increased risk of miscarriage.
Fatal Outcome
Ultimately, if Wilson’s disease is left untreated, the progressive damage to the liver and brain is fatal, typically before the age of 40.
Frequently Asked Questions (FAQs)
Who is at risk for Wilson disease?
The primary risk factor for Wilson’s disease is having a family history of the condition, specifically if parents or siblings are affected. Wilson’s disease is an autosomal recessive disorder. This means that a person must inherit two copies of the mutated ATP7B gene, one from each parent, to develop the disease.
At what age is Wilson’s disease usually diagnosed?
Wilson’s disease is typically diagnosed between the ages of 5 and 35 years old. The majority of individuals with Wilson’s disease develop symptoms and are diagnosed during their teenage years or early twenties. While less common, Wilson’s disease can also manifest and be diagnosed in older adults, even after the age of 40, and in rare cases, as late as the 60s or 70s. In these late-onset cases, neurological symptoms are often the predominant feature.
What can be mistaken for Wilson’s disease?
Wilson’s disease, with its variable presentation affecting the liver, brain, and other organs, can be mistaken for a number of other conditions. The differential diagnosis is broad and depends on the primary symptoms a patient presents with.
Liver-Related Presentations
- Autoimmune Hepatitis: Can present with similar liver inflammation, elevated liver enzymes, and even acute liver failure, particularly in young individuals. Sometimes, Wilson’s disease can initially be misdiagnosed as autoimmune hepatitis.
- Viral Hepatitis (A, B, C, D, E): Acute or chronic viral infections of the liver can cause jaundice, elevated liver enzymes, and fatigue, similar to the hepatic presentation of Wilson’s disease.
- Alcoholic Liver Disease: Chronic alcohol abuse can lead to liver damage, cirrhosis, and liver failure, mimicking the liver-related complications of Wilson’s disease.
- Non-Alcoholic Fatty Liver Disease (NAFLD) / Non-Alcoholic Steatohepatitis (NASH): These conditions can cause elevated liver enzymes and liver damage, although neurological symptoms and Kayser-Fleischer rings would be absent.
- Primary Biliary Cholangitis (PBC) and Primary Sclerosing Cholangitis (PSC): These autoimmune liver diseases can cause liver damage and elevated liver enzymes, but typically have different patterns of liver involvement and other distinguishing features.
- Drug-Induced Liver Injury: Certain medications can cause liver damage with symptoms similar to Wilson’s disease.
- Hemochromatosis: Another genetic disorder causing iron overload, which can lead to liver disease and cirrhosis. However, iron studies would be abnormal instead of copper studies.
- Alpha-1 Antitrypsin Deficiency: A genetic disorder that can cause liver disease and lung problems.
Neurological Presentations
- Other Movement Disorders: Conditions like Parkinson’s disease, Huntington’s disease, essential tremor, and other forms of dystonia can present with tremors, rigidity, and abnormal movements, potentially mimicking the neurological features of Wilson’s disease. However, these conditions usually lack the characteristic liver involvement and Kayser-Fleischer rings.
- Multiple Sclerosis (MS): Can cause neurological symptoms like incoordination, speech difficulties, and tremor, but typically involves white matter lesions on MRI and other distinct clinical features.
- Cerebellar Ataxia: Various genetic or acquired conditions can lead to problems with coordination and balance.
- Manganese Storage Disorders: These rare metabolic disorders can have neurological symptoms similar to Wilson’s disease.
- Leukodystrophies: A group of genetic disorders affecting the white matter of the brain, which can cause neurological symptoms.
Psychiatric Presentations
- Major Depressive Disorder, Anxiety Disorders, Schizophrenia, Bipolar Disorder: The psychiatric symptoms of Wilson’s disease can sometimes be the initial or prominent feature, leading to misdiagnosis as a primary psychiatric disorder. However, the presence of liver abnormalities or neurological signs should raise suspicion for Wilson’s disease.
Other Conditions
- Hemolytic Anemia (Coombs-negative): While hemolytic anemia can be a feature of Wilson’s disease, other conditions can also cause it, requiring further investigation to rule out Wilson’s disease.
- Congenital Disorders of Glycosylation (CDG): Some types of CDG can present with low serum copper and ceruloplasmin, mimicking Wilson’s disease.
- MEDNIK Syndrome: A rare genetic disorder affecting copper metabolism with neurological and other features that can overlap with Wilson’s disease.
- Aceruloplasminemia: A rare genetic disorder of iron metabolism that can have neurological symptoms and low ceruloplasmin, but with iron overload rather than copper overload.
- Menkes Disease and Occipital Horn Syndrome: These are X-linked disorders of copper deficiency due to mutations in ATP7A. While related to copper metabolism, they typically present with different clinical features and earlier in life than Wilson’s disease.
Is Wilson’s disease curable?
While there is currently no cure for Wilson’s disease, it is a very treatable condition. With proper and lifelong management, individuals with Wilson’s disease can often live normal or near-normal lifespans with a good quality of life.
What foods are high in copper?
For individuals with Wilson’s disease, it’s important to be mindful of foods that are naturally high in copper to help manage their copper levels, although medication is the primary treatment.
Very High in Copper
- Organ Meats: Especially liver (beef, calf, chicken) are exceptionally high in copper and should be avoided. Kidney and other organ meats are also high.
- Shellfish: Oysters are particularly high, as are other shellfish like clams, crab, lobster, shrimp, and mussels.
High in Copper
- Nuts: Cashews, Brazil nuts, almonds, walnuts, pecans, hazelnuts, and peanuts.
- Seeds: Sunflower seeds, sesame seeds, poppy seeds.
- Legumes: Soybeans, tofu, lentils, chickpeas, and dried beans (navy, pinto, white).
- Chocolate: Dark chocolate and cocoa powder are significant sources of copper. Milk chocolate contains less but should still be consumed in moderation.
- Mushrooms: Shiitake mushrooms have a particularly high copper content, but other varieties also contain copper.
- Dried Fruits: Raisins, apricots, figs, and other dried fruits can be relatively high in copper.
- Whole Grains: Wheat bran and whole-grain products.
- Spirulina: A type of blue-green algae often used as a dietary supplement.
- Molasses: A byproduct of sugar refining.
Moderate in Copper
- Avocados
- Sweet potatoes
- Potatoes (especially with the skin)
- Spinach and other dark leafy greens
- Many other vegetables and fruits contain smaller amounts of copper.
It’s important for individuals with Wilson’s disease to consult with their healthcare provider or a registered dietitian for personalized dietary guidance. The level of restriction needed can vary depending on the individual’s disease severity and treatment plan. The focus is usually on limiting very high-copper foods and consuming other foods in moderation while ensuring a balanced and nutritious diet.
Disclaimer: This article is intended for informational purposes only and is specifically targeted towards medical students. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Bandmann O, Weiss KH, Kaler SG. Wilson’s disease and other neurological copper disorders. Lancet Neurol. 2015 Jan;14(1):103-13. doi: 10.1016/S1474-4422(14)70190-5. PMID: 25496901; PMCID: PMC4336199.
- European Association for Study of Liver. EASL Clinical Practice Guidelines: Wilson’s disease. J Hepatol. 2012 Mar;56(3):671-85. doi: 10.1016/j.jhep.2011.11.007. PMID: 22340672.
- Shribman S, Marjot T, Sharif A, Vimalesvaran S, Ala A, Alexander G, Dhawan A, Dooley J, Gillett GT, Kelly D, McNeill A, Warner TT, Wheater V, Griffiths W, Bandmann O; British Association for the Study of the Liver Rare Diseases Special Interest Group. Investigation and management of Wilson’s disease: a practical guide from the British Association for the Study of the Liver. Lancet Gastroenterol Hepatol. 2022 Jun;7(6):560-575. doi: 10.1016/S2468-1253(22)00004-8. Epub 2022 Apr 13. PMID: 35429442.
- Mulligan C, Bronstein JM. Wilson Disease: An Overview and Approach to Management. Neurol Clin. 2020 May;38(2):417-432. doi: 10.1016/j.ncl.2020.01.005. Epub 2020 Feb 28. PMID: 32279718.
- Hermann, W. Classification and differential diagnosis of Wilson’s disease. Annals of Translational Medicine. 2019; 2(7).