TL;DR
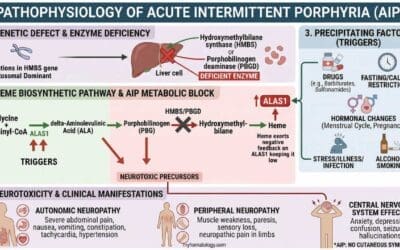
Beta thalassemia is a hereditary hemoglobin disorder due to reduced or absence of beta-globin chain production leading to ineffective erythropoiesis and anemia. It is predominantly an autosomal recessive gene disorder.
Classifications
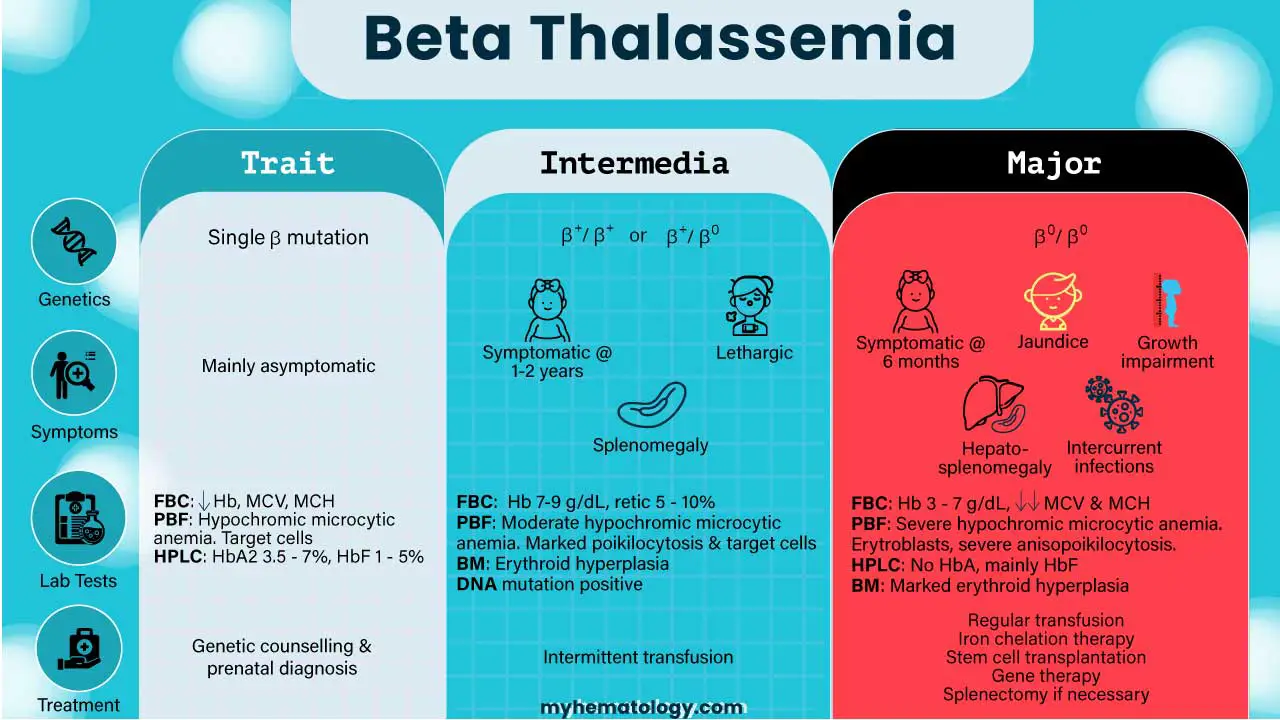
| Characteristic | Beta Thalassemia Minor (Trait) | Beta Thalassemia Intermedia | Beta Thalassemia Major (Cooley’s Anemia) |
| Genetic basis | Single β mutation | β+/ β+ or β+/ β0 or severe mutations with co-inheritance of alpha thalassemia or hereditary persistence of fetal hemoglobin (HPFH). | β0/ β0 |
| Severity | Mildest form. | Milder than major, but more severe than minor. | Most severe form, also known as Cooley’s Anemia. |
| Hemoglobin Production | Minimal quantitative reduction in beta globin chains; alpha/beta chain ratio is almost balanced. | Low beta chain production leading to excess unpaired alpha chains, causing ineffective erythropoiesis and moderate anemia. | No beta globin chain production, leading to severe excess of unpaired alpha globin chains, ineffective erythropoiesis, and extravascular hemolysis. |
| Typical Onset | Often detected incidentally through routine blood tests; individuals are typically asymptomatic. | Signs and symptoms typically appear in early childhood or later in life, often around 2-6 years of age. | Infants are typically normal at birth but develop severe, life-threatening anemia during the first year of life, usually around 3-6 months of age |
| Need for Transfusions | Generally no treatment required and no need for transfusions. | Generally non-transfusion-dependent, but may require intermittent transfusions during periods of stress or severe anemia. | Transfusion-dependent, requiring frequent, lifelong regular blood transfusions for survival. |
| Common Symptoms ▾ | Mainly asymptomatic; may have small red blood cells (microcytic) and sometimes mild anemia. | Mild to moderate anemia, pallor, enlarged spleen (splenomegaly), slowed growth, bone abnormalities, increased risk of abnormal blood clots | Severe anemia, failure to thrive, fatigue, weakness, jaundice, bone deformities (e.g., “chipmunk face,” frontal bossing), enlarged spleen and liver, enlarged heart, intercurrent infections, features of iron overload, delayed puberty, short stature. |
| Prognosis/Life Expectancy (with treatment for severe forms) | Normal life expectancy. | Can live relatively normal lives with proper medical management; life expectancy varies widely. | Historically, early death (often within the first decade or by ages 20-30 due to heart failure); with advancements in regular blood transfusions and iron chelation therapy, many individuals now live into their 50s and 60s. |
*Click ▾ for more information
What is beta thalassemia?
Beta thalassemia is a genetic blood disorder that reduces the production of hemoglobin in the red blood cells. Thalassemia is defined as the quantitative reduction of globin chains production leading to anemia.
Thalassemia can be loosely classified into alpha thalassemia and beta thalassemia depending on the affected globin gene. Beta thalassemia occurs due to a mutation found in the beta globin gene or the promoter region which leads to a quantitative reduction or absence of the beta globin chain production.
Even though the production of the beta globin chain has decreased, the production of alpha globin chains is not affected and produces normally. These will lead to a lack of Hb A formation as well as an excess of free alpha globin chains. Free alpha globin chains precipitate as monomers and cause membrane damage leading to anemia as well as an increase in oxidative reactive oxygen species and thus oxidative stress in the cell.
What is the function of the beta-globin gene in hemoglobin?
The beta-globin gene is a gene that provides instructions for making a protein called beta-globin. Beta-globin is a component (subunit) of a larger protein called hemoglobin, which is located inside red blood cells. Hemoglobin is responsible for carrying oxygen from the lungs to the body’s tissues and carbon dioxide from the tissues back to the lungs.
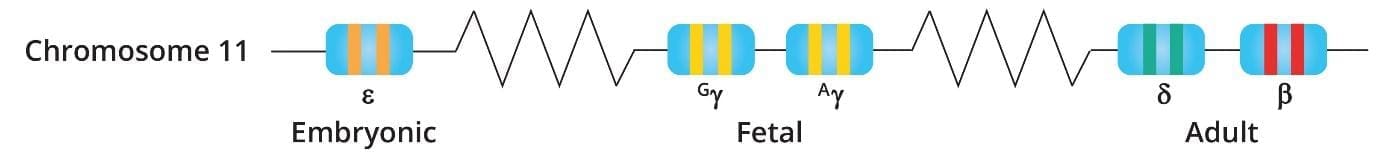
The beta-globin gene is located on chromosome 11. Humans have two copies of the beta-globin gene, one on each chromosome 11. In adults, hemoglobin consists of four protein subunits: usually two subunits of beta-globin and two subunits of a protein called alpha-globin, which is produced from another gene called HBA.
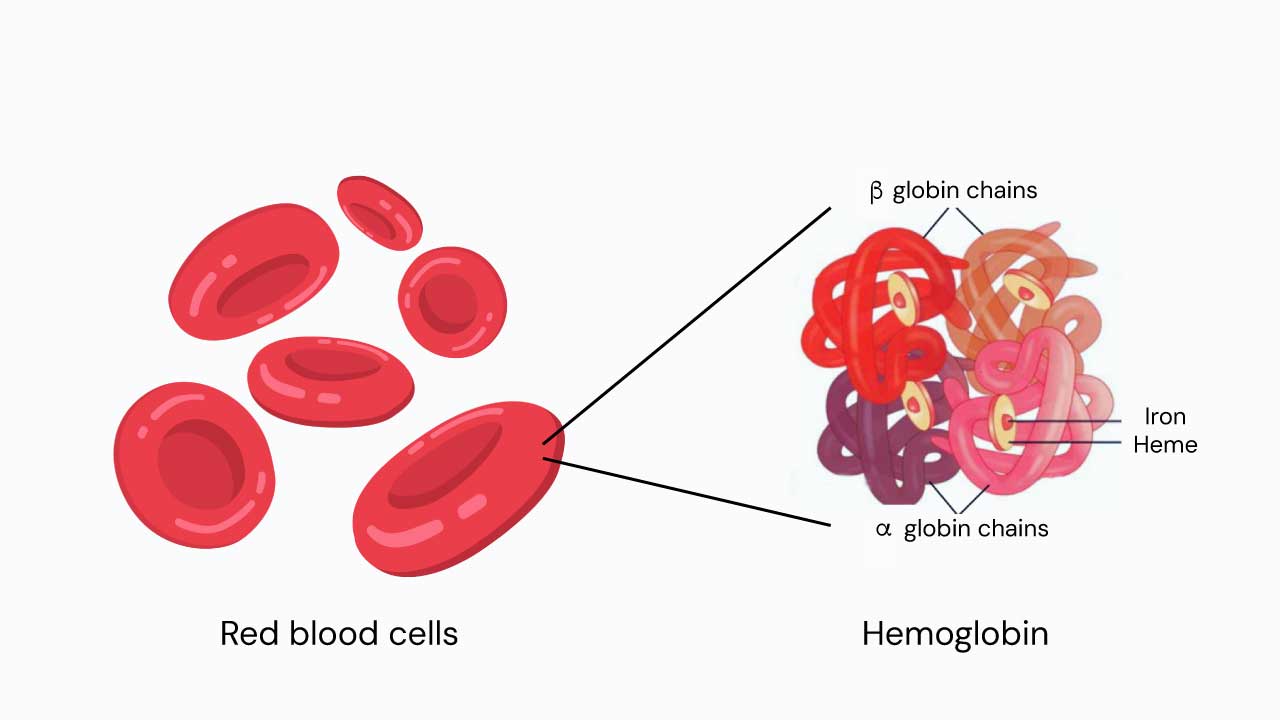
The beta-globin gene is responsible for producing the beta-globin subunit of hemoglobin. The beta-globin subunit has a pocket that can bind to an iron-containing molecule called heme. Hemoglobin can bind up to four oxygen molecules, one to each heme molecule.
When red blood cells reach the lungs, oxygen binds to the beta-globin subunits of hemoglobin. The red blood cells then travel throughout the body, delivering oxygen to the tissues. When the red blood cells reach the tissues, oxygen is released from the beta-globin subunits of hemoglobin and diffuses into the tissues.
Carbon dioxide is a waste product of metabolism. It is produced in the tissues and diffuses into the red blood cells. In the red blood cells, carbon dioxide binds to the beta-globin subunits of hemoglobin. The red blood cells then travel back to the lungs, where the carbon dioxide is released and exhaled.
The beta-globin gene is essential for life. Without the beta-globin gene, humans would not be able to produce hemoglobin and would not be able to carry oxygen to the tissues. Mutations in the beta-globin gene can lead to a variety of blood disorders, including beta thalassemia and sickle cell anemia.
How does beta thalassemia occur?
Beta thalassemia is due to a mutation in the beta-globin gene. Mutations in the beta-globin gene can reduce or eliminate the production of beta-globin. This can lead to a deficiency of hemoglobin causing anemia. The severity of this disorder depends on the severity of the mutation in the beta-globin gene.
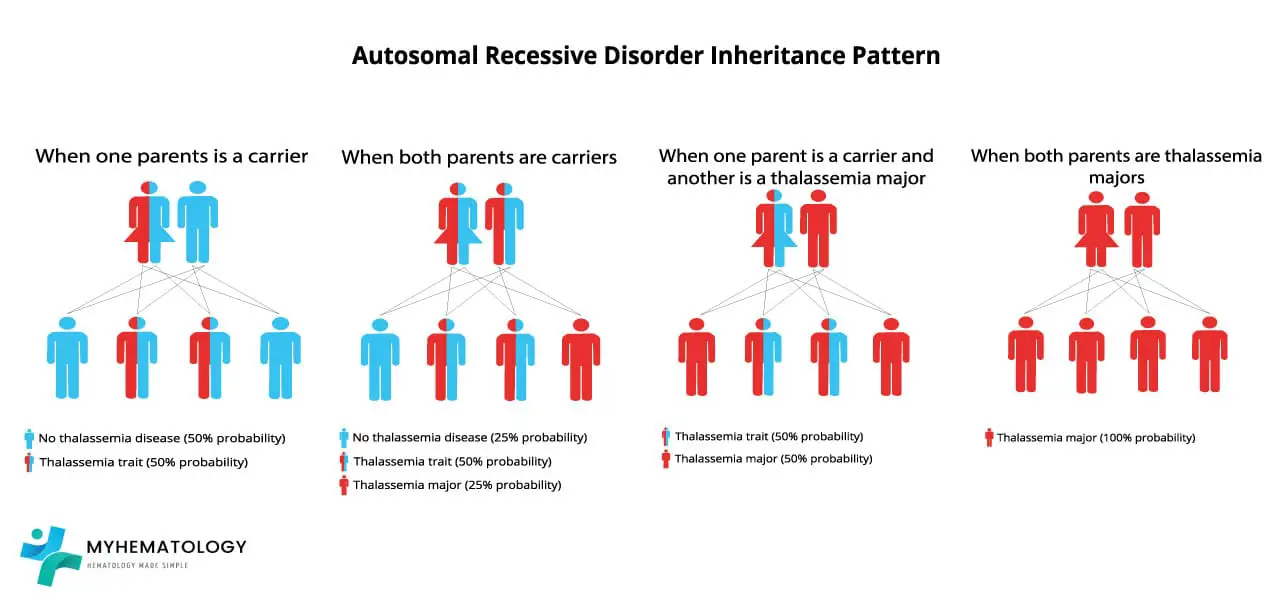
Beta thalassemia is an autosomal recessive inherited disorder, meaning that it is passed down from parents to children.
If one parent is a carrier of the beta-globin gene mutation, there is a 50% probability that the offspring will inherit the mutation. If both parents are carriers of the beta-globin gene mutation, there is a 25% probability that the offispring will have beta thalassemia major, a 50% probability that the offspring will be carriers, and a 25% probability that the offspring will not have the mutant allele at all.
The core of beta thalassemia’s pathophysiology is the mismatch in the production of globin chains. In beta thalassemia, a genetic mutation on chromosome 11 affects the HBB gene, causing a deficit in beta-globin production. Since the alpha-globin genes are unaffected, there’s a relative excess of alpha-globin chains. These unpaired alpha-globin chains are highly unstable and toxic, leading to the key problems of the disease.
The excess alpha-globin chains don’t form functional hemoglobin. Instead, they precipitate and form inclusion bodies within red blood cell precursors in the bone marrow. This causes two major issues:
- Ineffective Erythropoiesis: The presence of these toxic inclusions damages the red blood cell precursors, leading to their premature death in the bone marrow. This means that a large number of red blood cells are destroyed before they even mature and enter the bloodstream. The body tries to compensate for this by ramping up red blood cell production, causing the bone marrow to expand. This expansion can lead to bone deformities and osteoporosis.
- Hemolysis: The few red blood cells that do manage to mature and escape the bone marrow still contain the toxic alpha-globin precipitates. These cells have fragile membranes and a shortened lifespan, leading to their rapid destruction in the spleen. This process is called hemolysis. The premature destruction of red blood cells contributes significantly to the severe anemia seen in patients.
The combination of ineffective erythropoiesis and hemolysis leads to several systemic consequences:
- Anemia: Both the destruction of immature red blood cells and the shortened lifespan of mature ones result in a severe, chronic anemia, which is the hallmark of the disease.
- Splenomegaly and Extramedullary Hematopoiesis: To cope with the anemia, the body activates alternative sites for blood cell production (extramedullary hematopoiesis), particularly in the liver and spleen. This can lead to the enlargement of these organs (hepatosplenomegaly). The spleen also becomes enlarged because it’s working overtime to clear the damaged red blood cells from circulation.
- Iron Overload: This is a major complication. The body’s attempt to produce more red blood cells inappropriately increases iron absorption from the gut. Additionally, many patients require frequent blood transfusions, which are a major source of iron. Without proper management, this excess iron accumulates in vital organs like the heart, liver, and endocrine glands, leading to organ damage and failure.
Epidemiology
This disorder is most common in people of Mediterranean, Middle Eastern, and Southeast Asian descent. However, it can occur in people of any ethnicity.
Molecular Classifications
The molecular classifications of beta thalassemia can be loosely classified into
- Beta thalassemia minor (trait): Usually represents the inheritance of a single beta-thalassemia allele and the hemoglobin reduction is mild.
- Beta thalassemia intermedia: Could involve homozygosity of compound heterozygosity for mild mutations or homozygosity or compound heterozygosity for severe mutations with the co-inheritance of alpha thalassemia or heterocellular hereditary persistence of foetal hemoglobin (HPFH).
- Beta thalassemia major: Usually represents the inheritance of homozygosity or compound heterozygosity for severe mutations.
Signs & Symptoms
The clinical features of beta thalassemia vary depending on the severity of the condition.
| Type of Beta Thalassemia | Signs and Symptoms |
| Beta Thalassemia Minor (Trait) | – Mainly asymptomatic. – Often detected incidentally through routine blood tests. – May have small red blood cells (microcytic). – Can sometimes present with mild anemia. |
| Beta Thalassemia Intermedia | – Signs and symptoms typically appear in early childhood or later in life. – Mild to moderate anemia. – Pallor (paleness). – Enlarged spleen (splenomegaly). – Bone abnormalities or deformities. – Increased risk of developing abnormal blood clots. – Growth and development may be retarded, even if able to survive without regular blood transfusions. – Symptoms are highly variable depending on various genetic modifiers. |
| Beta Thalassemia Major | – Most severe form, also known as Cooley’s Anemia. – Infants are typically normal at birth but develop severe, life-threatening anemia during the first year of life, usually around 3-6 months of age. – Failure to thrive (difficulty gaining weight and growing at the expected rate). – Fatigue and weakness. – Yellowing of the skin and whites of the eyes (jaundice). – Shortness of breath. – Poor appetite. – Bone deformities, particularly in the face (frontal bossing, “thalassemia facies” or “chipmunk face”), skull, and long bones. Bones can widen, become thin and brittle, increasing fracture risk. – Enlarged spleen (splenomegaly) and liver (hepatomegaly). – Intercurrent infections. – Features of iron overload, which can lead to complications such as heart failure, liver cirrhosis, and endocrine problems like diabetes mellitus, hypopituitarism, hypothyroidism, and hypoparathyroidism. – Delayed puberty and secondary sexual characteristics. – Osteoporosis and short stature. – High red cell turnover can predispose to gallstone formation. – Extramedullary hematopoiesis. – Without treatment, individuals typically die within the first decade of life, or even within the first two years. Severe thalassemia can cause early death (between ages 20 and 30) due to heart failure. |
Complications
Beta thalassemia can lead to a range of complications, particularly in its more severe forms (intermedia and major), due to the body’s inability to produce sufficient functional hemoglobin and the resulting issues with red blood cell production and iron regulation. These complications can affect multiple organ systems and significantly impact a patient’s quality of life and prognosis.
- Iron Overload (Hemosiderosis): This is one of the most significant complications, especially in individuals requiring regular blood transfusions. Each unit of transfused blood contains iron, and the body has no natural way to excrete excess iron. Even without transfusions, ineffective erythropoiesis (the premature destruction of red blood cell precursors in the bone marrow) can lead to increased intestinal iron absorption. This excess iron accumulates in various organs, including the heart, liver, and endocrine glands, leading to severe damage and dysfunction.
- Heart Problems: Iron deposition in the heart can cause cardiomyopathy (weakening of the heart muscle), congestive heart failure, and irregular heart rhythms (arrhythmias), which are major causes of early death in severe thalassemia.
- Liver Problems: Iron overload can lead to liver cirrhosis and liver failure.
- Endocrine Problems: Iron accumulation can damage endocrine glands, resulting in:
- Diabetes Mellitus: Due to damage to the pancreas.
- Hypopituitarism: Affecting the pituitary gland, which can lead to growth retardation and delayed puberty.
- Hypothyroidism: Affecting the thyroid gland.
- Hypoparathyroidism: Affecting the parathyroid glands.
- Delayed Puberty and Secondary Sexual Characteristics: Often seen in adolescents with thalassemia major.
- Bone Changes and Deformities: The severe anemia in beta thalassemia triggers the bone marrow to expand in an attempt to produce more red blood cells. This expansion can cause bones to widen and become thin and brittle, increasing the risk of fractures. Characteristic bone deformities, particularly in the face (e.g., “thalassemia facies” or “chipmunk face” with frontal bossing), and skull (“hair-on-end” appearance on X-rays), can develop. Osteoporosis and short stature are also common.
- Enlarged Spleen (Splenomegaly) and Liver (Hepatomegaly): The spleen works harder to remove old or damaged red blood cells, leading to its enlargement. An enlarged spleen can worsen anemia by trapping and destroying even more red blood cells, including those from transfusions. The liver may also enlarge due to extramedullary hematopoiesis (blood cell production outside the bone marrow) and iron overload.
- Slowed Growth and Development: Chronic anemia and iron overload can significantly slow a child’s growth and delay puberty.
- Increased Risk of Infections: Individuals with thalassemia, especially those who undergo splenectomy (surgical removal of the spleen) to manage splenomegaly, are at a higher risk of severe infections.
- Abnormal Blood Clots (Thrombosis): People with beta thalassemia, particularly intermedia, are at an increased risk of developing abnormal blood clots. This can lead to serious conditions like pulmonary hypertension and embolism.
- Gallstone Formation: The high turnover rate of red blood cells can predispose individuals to the formation of gallstones.
- Extramedullary Hematopoiesis: In severe cases, the body attempts to produce red blood cells in organs outside the bone marrow, such as the spleen and liver, leading to their enlargement and potential dysfunction.
Without proper treatment, severe beta thalassemia (major) can be life-threatening, with individuals historically dying within the first decade of life, often due to heart failure. However, advancements in medical care, including regular blood transfusions and iron chelation therapy, have significantly improved the prognosis, allowing many individuals to live into their 50s and 60s
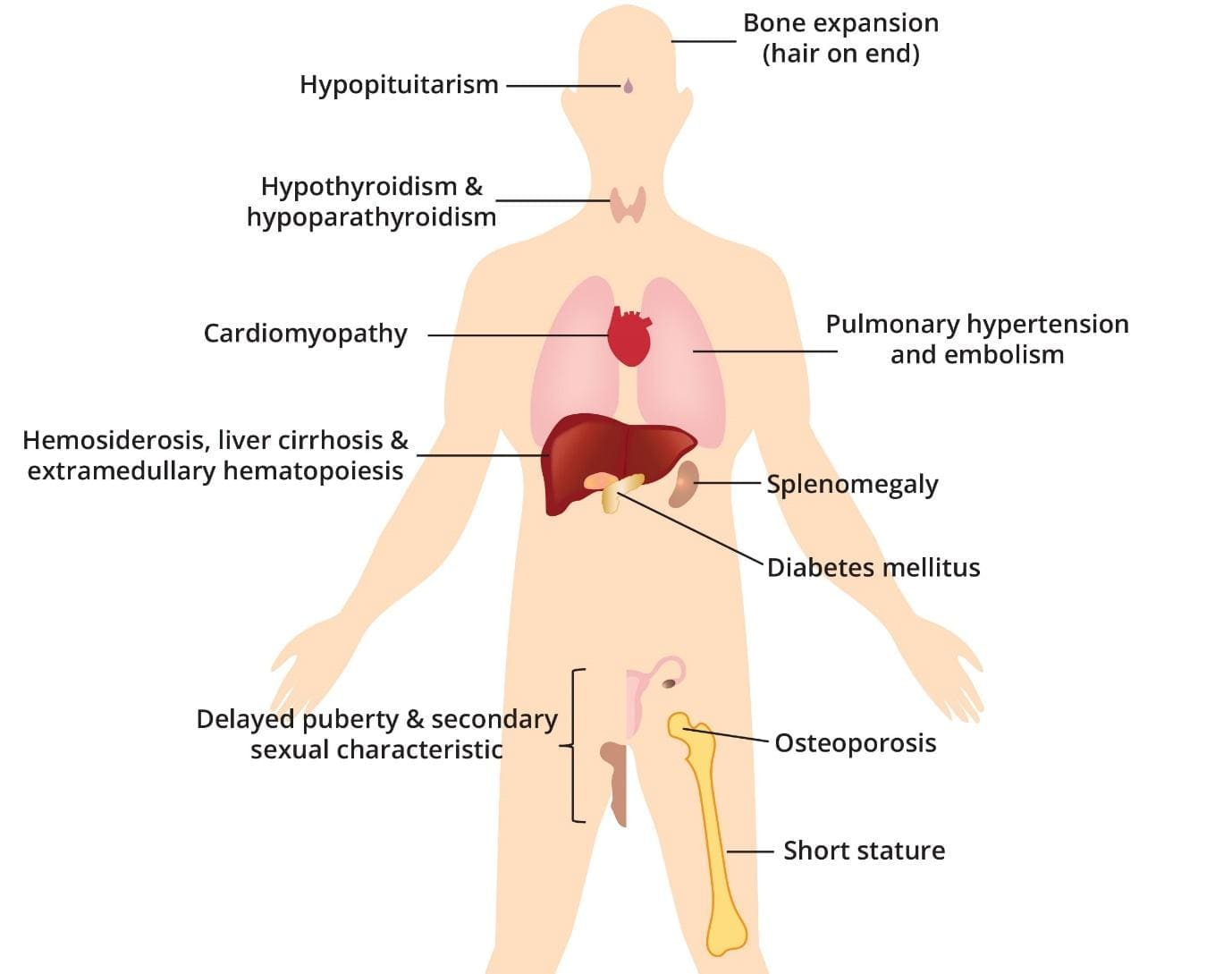
Beta thalassemia major results in the abnormal production of hemoglobin, leading to chronic anemia. The repeated blood transfusions required to manage anemia introduce excess iron into the body, gradually accumulating in various organs.
Laboratory investigations
Laboratory investigations play an important role in the diagnosis and management of beta thalassemia.
Complete blood count (CBC)
A CBC is a test that measures the number of different types of blood cells in the blood. A low red blood cell count and a low hemoglobin level are suggestive of anemia. Beta thalassemia is characterised by low hemoglobin level, low MCV and MCH and increased reticulocyte count.
Beta thalassemia minor (trait) has hypochromic microcytic red blood cells with or without a mild anemia, increased levels of Hb A2 and variable increases in Hb F levels.
Peripheral blood smear
A peripheral blood smear is a microscopic examination of blood cells. It can be used to look for signs of anemia, such as small, pale red blood cells.
- In beta thalassemia minor (trait), the peripheral blood smear shows hypochromic microcytic red cells with presence of target cells.
- Beta thalassemia intermedia will have moderate microcytic hypochromic anemia with reticulocytosis.
- Beta thalassemia major will show marked anisopoikilocytosis with numerous target cells and nucleated red cells may be seen.
Hemoglobin electrophoresis
Hemoglobin electrophoresis is a test that is used to identify the different types of hemoglobin in the blood. In beta thalassemia, hemoglobin electrophoresis may show a decrease in the amount of normal hemoglobin (HbA) and an increase in the amount of abnormal hemoglobin (such as HbS, HbE, or HbF).
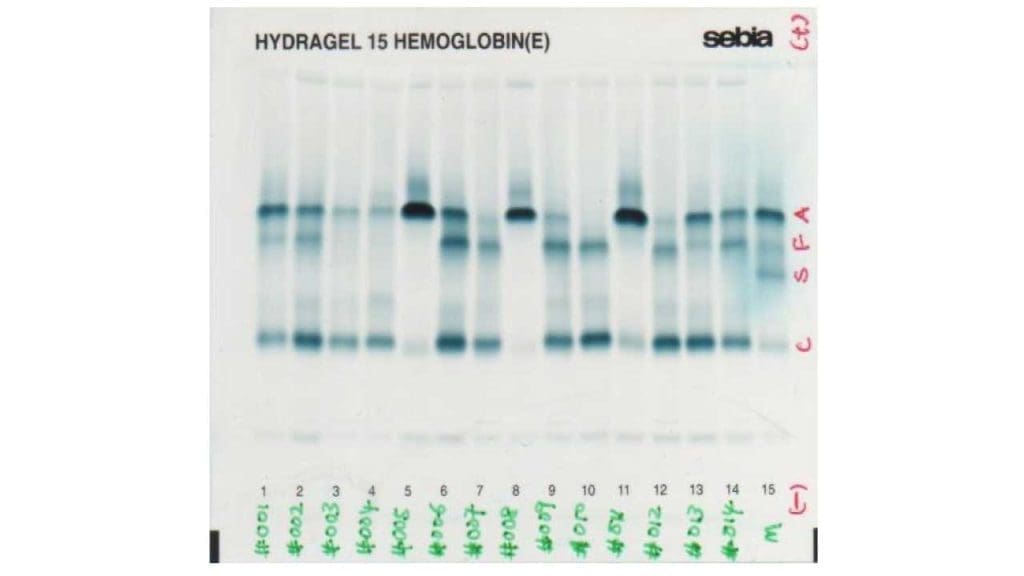
High performance liquid chromatography (HPLC)
Similar to hemoglobin electrophoresis, there will be a quantitative reduction or absence of Hb A with an increase of Hb A2 and Hb F.
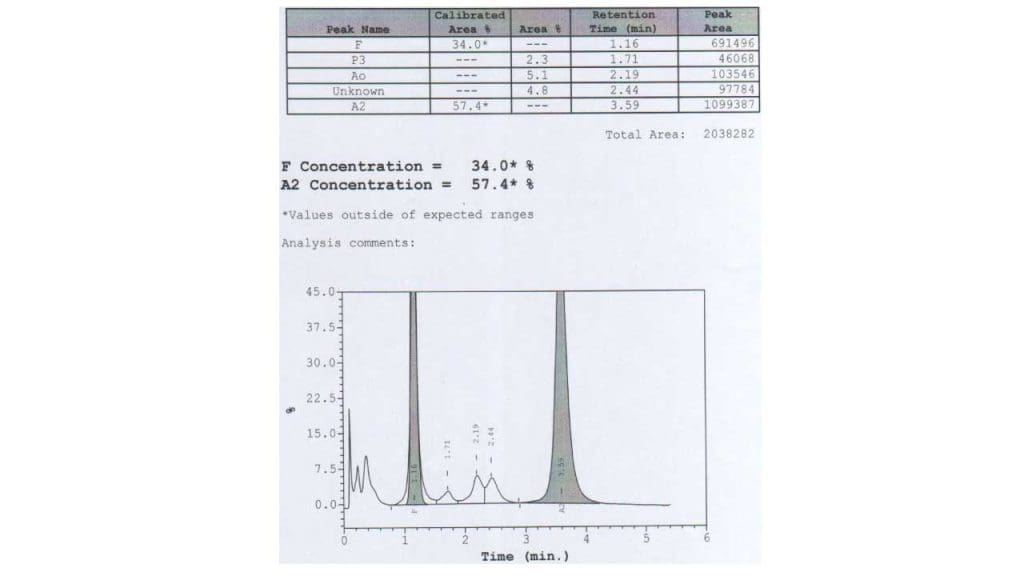
DNA testing
DNA testing is the most definitive test for the diagnosis of beta thalassemia. It can be used to identify the specific mutation in the beta-globin gene that is causing beta thalassemia. DNA testing for example PCR can also be used to identify carriers of beta thalassemia, which is important for genetic counseling and prenatal diagnosis.
Other laboratory tests
Other laboratory tests that may be used to investigate beta thalassemia include:
- Iron studies: Iron studies are used to measure the amount of iron in the blood and body tissues. Beta thalassemia major individuals are at risk for iron overload due to the regular blood transfusions that they require. There will be a marked increase in serum iron, serum ferritin and transferrin saturation level while low TIBC.
- Liver function tests: Liver function tests are used to assess the function of the liver. Beta thalassemia major individuals are at risk for liver damage due to iron overload.
- Cardiac function tests: Cardiac function tests are used to assess the function of the heart. Beta thalassemia major individuals are at risk for heart problems due to iron overload, anemia, and other complications of the disorder.
- Other investigations will show increased bilirubin and urobilinogen level with decreased serum haptoglobin.
How is beta thalassemia treated?
The treatment and management of this disorder vary significantly depending on the severity of the condition, ranging from no treatment for mild forms to complex, lifelong interventions for severe cases. The primary goals of management are to address anemia, prevent complications from iron overload, and improve the patient’s quality of life.
Management by Thalassemia Type
Beta Thalassemia Minor (Trait)
Individuals with beta thalassemia minor are typically asymptomatic and generally do not require any specific treatment.
Genetic counseling is highly recommended for carriers, especially if they are considering having children, to understand the risk of passing on the condition to their offspring.
Beta Thalassemia Intermedia
This form is milder than beta thalassemia major, and individuals may or may not require regular blood transfusions. Intermittent transfusions may be given as needed, particularly during periods of increased stress, illness, or if anemia becomes severe.
Iron chelation therapy may be used to prevent iron overload, especially as patients get older, even if they don’t receive regular transfusions, due to increased intestinal iron absorption.
Beta Thalassemia Major (Transfusion-Dependent Thalassemia)
This is the most severe form and requires aggressive, lifelong management. Without proper treatment, individuals typically do not survive past the first decade of life.
Key Treatments for Severe Forms (Beta Thalassemia Major)
Regular Blood Transfusions
This is the first-line and cornerstone treatment for beta thalassemia major.
Patients receive packed red cell transfusions typically every 2 to 4 weeks to maintain adequate hemoglobin levels, alleviate severe anemia, and suppress ineffective erythropoiesis.
Appropriate transfusions help children grow well and can significantly improve life expectancy, allowing many individuals to live into their 50s and 60s.
Iron Chelation Therapy
A critical component of management for transfusion-dependent patients. Regular blood transfusions introduce excess iron into the body, leading to iron overload (hemosiderosis), which can damage vital organs like the heart, liver, and endocrine glands.
Chelation therapy involves medications (chelating agents) that bind to excess iron in the body, allowing it to be excreted. This prevents or reduces organ damage and improves prognosis.
Even in non-transfusion-dependent forms, iron overload can occur due to increased intestinal iron absorption from ineffective erythropoiesis, necessitating chelation therapy.
Splenectomy (Surgical Removal of the Spleen)
The spleen often becomes enlarged (splenomegaly) in thalassemia due to its increased role in destroying abnormal red blood cells.
Splenectomy may be indicated if there is significant splenomegaly leading to excessive destruction of transfused red cells and increased transfusion requirements. However, splenectomy carries risks, including a higher risk of thrombotic complications (blood clots) and post-splenectomy sepsis, so it is generally recommended only in children older than 6 years or when absolutely necessary.
Bone Marrow Transplantation (Hematopoietic Stem Cell Transplantation)
Currently, bone marrow transplantation is considered the only potential cure for beta thalassemia. It involves replacing the patient’s faulty bone marrow with healthy stem cells from a compatible donor.
This treatment is primarily offered to children and can eliminate the need for lifelong blood transfusions.
Gene Therapy
Gene therapy has shown remarkable progress and is considered a promising area for a potential cure.
Novel gene therapies, such as CASGEVY (exagamglogene autotemcel) and Betibeglogene Autotemcel, are being used to treat beta thalassemia.
These are often one-time treatments that modify a patient’s own blood-forming stem cells, enabling them to produce more healthy hemoglobin and potentially reduce or eliminate the need for ongoing blood transfusions. CASGEVY, for example, uses CRISPR/Cas9 gene editing technology.
Supportive Care and Important Considerations
- Monitoring for Complications: Ongoing monitoring for complications such as heart failure, liver problems, endocrine issues (e.g., diabetes, delayed puberty), bone deformities, and infections is essential for comprehensive management.
- Folate Supplements: Folate (folic acid) supplements are often given to support red blood cell production, as the body’s demand for red blood cells is high in thalassemia.
- Avoidance of Iron Supplements: It is crucial that individuals with thalassemia, especially those receiving transfusions, do not take iron supplements unless specifically advised by a doctor. Their bodies already accumulate too much iron, and additional iron can worsen iron overload and organ damage.
- Genetic Counseling: For families with a history of thalassemia, genetic counseling is vital for understanding inheritance patterns, carrier status, and reproductive options, including prenatal diagnosis. Many countries with high frequency of beta thalassemia run a prevention screening program while offering genetic counselling to those affected.
Transfusion-Dependent Thalassemia (TDT)
The classification of beta thalassemia into “transfusion-dependent” and “non-transfusion-dependent” types primarily distinguishes the severity of the condition and the necessity of regular blood transfusions for survival and management
- Severity: This is the most severe form of beta thalassemia, often referred to as Beta Thalassemia Major or Cooley’s Anemia.
- Onset of Symptoms: Infants with this type are typically healthy at birth but develop severe, life-threatening anemia within the first year of life, usually around 3 to 6 months of age.
- Need for Transfusions: Individuals with TDT have such severe symptoms that they require frequent, often lifelong, regular blood transfusions to replenish their red blood cell supply and manage their anemia. Without these regular transfusions and proper treatment, individuals with beta thalassemia major typically do not survive past the first decade of life, or even the first two years.
- Genetic Basis: It usually results from inheriting two severe beta thalassemia mutations (homozygosity or compound heterozygosity for severe mutations).
- Complications: Due to the chronic transfusions, iron overload (hemosiderosis) is a major concern, leading to severe complications affecting the heart, liver, and endocrine glands.
Non-Transfusion-Dependent Thalassemia (NTDT)
- Severity: This type is milder than transfusion-dependent thalassemia. It encompasses Beta Thalassemia Intermedia and, in some classifications, Beta Thalassemia Minor (Trait).
- Onset of Symptoms: Symptoms for beta thalassemia intermedia typically appear in early childhood or later in life. Beta thalassemia minor individuals are often asymptomatic and may only be detected incidentally.
- Need for Transfusions: Individuals with NTDT generally do not require regular, lifelong blood transfusions for survival. While they may experience mild to moderate anemia, they can often manage without consistent transfusions. Intermittent transfusions might be needed in specific situations, such as during periods of increased stress or illness.
- Genetic Basis: Beta thalassemia intermedia can involve homozygosity or compound heterozygosity for milder beta thalassemia mutations, or severe mutations co-inherited with other genetic modifiers that lessen the severity. Beta thalassemia minor occurs when an individual inherits a variant gene from only one parent, making them a carrier, usually with mild or no symptoms.
- Complications: While generally milder, individuals with NTDT can still develop complications, including mild to moderate anemia, slow growth, bone abnormalities, and an increased risk of developing abnormal blood clots. Iron overload can still occur, even without transfusions, due to increased intestinal iron absorption.
Frequently Asked Questions (FAQs)
What is the life expectancy of a person with beta thalassemia?
The life expectancy of a person with beta thalassemia significantly depends on the severity of the condition and the availability of proper medical care.
Types of Beta Thalassemia and Life Expectancy
- Beat thalassemia minor (trait): People with this mild form typically have a normal life expectancy.
- Beta thalassemia intermedia: Life expectancy can vary widely depending on the severity of symptoms. With proper medical management, many individuals can live relatively normal lives.
- Beta thalassemia major: Historically, life expectancy was significantly shortened. However, with advancements in medical treatment, including regular blood transfusions and iron chelation therapy, many individuals with beta thalassemia major are now living into their 50s and 60s.
Does beta thalassemia get worse with age?
Beta thalassemia itself doesn’t necessarily worsen with age. However, the risk of complications associated with the condition does increase over time.
Common Complications
- Iron overload: This is a major concern due to regular blood transfusions.
- Bone problems: These can develop due to the body’s attempt to produce more red blood cells.
- Liver and heart issues: These can be caused by iron overload.
- Blood clots: Increased risk due to changes in blood composition.
Can people with thalassemia donate blood?
Generally, individuals with beta thalassemia intermedia or major cannot donate blood. Thalassemia is a blood disorder that affects the production of hemoglobin, the protein in red blood cells that carries oxygen. People with thalassemia intermedia and major often rely on regular blood transfusions to survive, making them ineligible to donate. Additionally, their blood may not meet the necessary health standards for donation.
How to check for beta thalassemia?
To check for beta thalassemia, a blood test is typically performed. This involves a complete blood count (CBC) to examine red blood cells, and hemoglobin electrophoresis to analyze the types of hemoglobin present. In some cases, genetic testing may also be necessary to confirm the diagnosis.
Disclaimer: This article is intended for informational purposes only and is specifically targeted towards medical students. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Weatherall D. 2003 William Allan Award address. The Thalassemias: the role of molecular genetics in an evolving global health problem. Am J Hum Genet. 2004 Mar;74(3):385-92. doi: 10.1086/381402. PMID: 15053011; PMCID: PMC1182250.
- Steinberg MH, Forget BG, Higgs DR, Weatherall DJ. Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management (Cambridge Medicine) 2nd Edition. 2009.
- Weatherall D. Thalassaemia: The Biography (Biographies of Disease)(OUP Oxford). 2010.
- Needs T, Gonzalez-Mosquera LF, Lynch DT. Beta Thalassemia. [Updated 2023 May 1]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK531481/
- Langer AL. Beta-Thalassemia. 2000 Sep 28 [Updated 2024 Feb 8]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1426/
- Galanello, R., Origa, R. Beta-thalassemia. Orphanet J Rare Dis 5, 11 (2010). https://doi.org/10.1186/1750-1172-5-11
- Natesirinilkul, R. (2020). Thromboembolism in Beta-Thalassemia Disease. IntechOpen. doi: 10.5772/intechopen.89313