TL;DR
Dyskeratosis congenita is a rare, inherited bone marrow failure syndrome caused by defects in telomere maintenance. This leads to abnormally short telomeres and premature cellular aging, primarily affecting rapidly dividing cells.
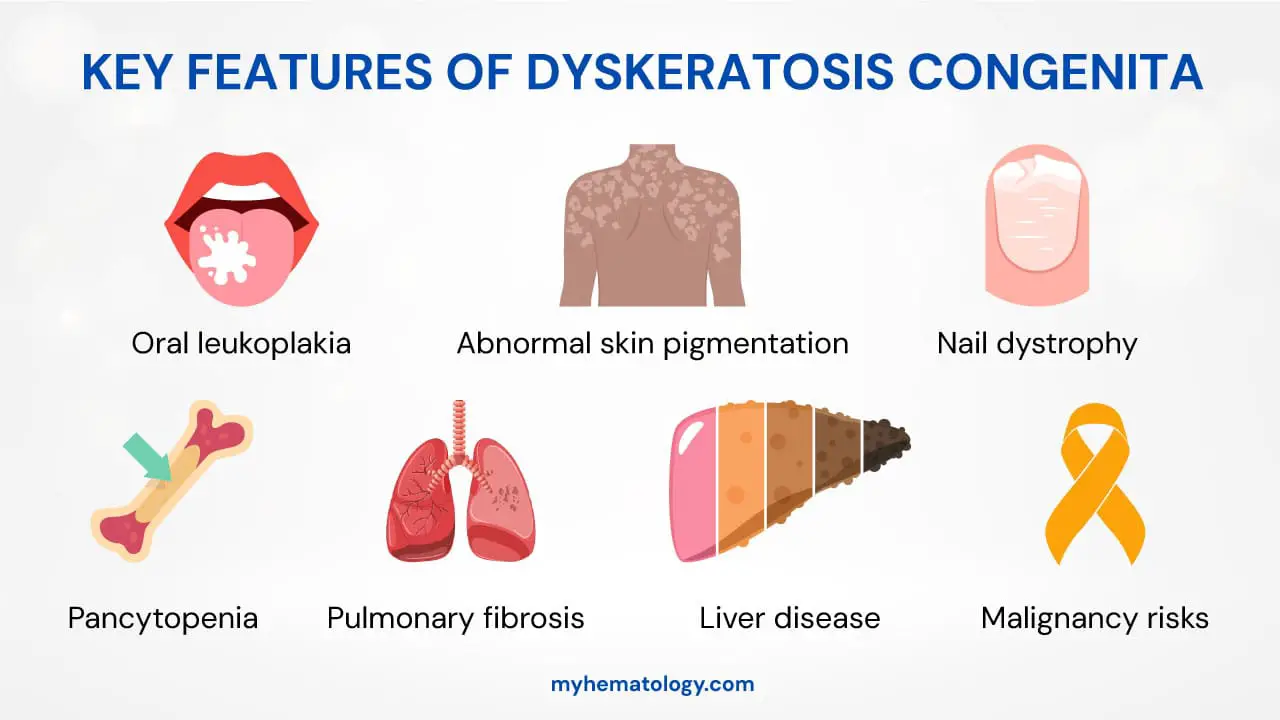
Key Clinical Manifestations ▾:
- Classic Mucocutaneous Triad:
- Nail Dystrophy (brittle, ridged, or absent nails).
- Abnormal Skin Pigmentation (a “lacy,” mottled appearance).
- Oral Leukoplakia (white patches in the mouth with a risk of cancer).
- Systemic Complications: Progressive bone marrow failure is the leading cause of death. Other serious issues include pulmonary fibrosis and a significantly increased risk of malignancies (e.g., squamous cell carcinoma, MDS, AML).
- Clinical suspicion is based on the mucocutaneous triad and symptoms of bone marrow failure.
- Definitive diagnosis requires demonstrating critically short telomeres through specialized lab tests (e.g., quantitative PCR, Flow-FISH).
- Genetic testing confirms the diagnosis by identifying a mutation in a telomere-related gene.
- Supportive Care: Includes blood transfusions and hematopoietic growth factors to manage cytopenias.
- Curative Treatment: Hematopoietic Stem Cell Transplantation (HSCT) is the only curative option for bone marrow failure, but it is a high-risk procedure.
- Monitoring: Long-term surveillance is crucial for detecting and managing complications like cancer and organ damage.
*Click ▾ for more information
Dyskeratosis congenita (DC), also known as Zinsser-Engman-Cole syndrome, is a rare inherited disorder characterized by a classic triad of clinical features: abnormal skin pigmentation (reticulated hyperpigmentation), nail dystrophy, and oral leukoplakia. However, its most severe and life-threatening manifestations are due to the underlying defect in telomere maintenance, leading to premature aging and a wide range of systemic complications, most notably progressive bone marrow failure.
As a telomere biology disorder (TBD), understanding Dyskeratosis congenita (DC) is critical for allied health professionals to facilitate early diagnosis and intervention, significantly impacting patient outcomes.
Causes and Pathophysiology
At the core of dyskeratosis congenita’s pathology lies a defect in the telomere-telomerase complex. Telomeres are protective caps at the ends of chromosomes, composed of repetitive DNA sequences (TTAGGG) and a complex of associated proteins known as shelterin. They are essential for maintaining genomic integrity and preventing chromosomal fusion and degradation. With each cell division, telomeres naturally shorten. Telomerase, a reverse transcriptase enzyme, counteracts this shortening by adding new telomeric DNA.
In dyskeratosis congenita, mutations in genes encoding components of the telomerase complex or telomere-associated proteins lead to critically short telomeres. These mutations can be inherited in X-linked, autosomal dominant, or autosomal recessive patterns. Over 15 genes have been identified, including DKC1 (the most common, X-linked), TERT (autosomal dominant), and TERC (autosomal dominant). Others include NOP10 and NHP2 (autosomal recessive).
Short telomeres trigger a DNA damage response, leading to cellular senescence (premature aging) and apoptosis. This process disproportionately affects highly proliferative tissues, such as hematopoietic stem cells in the bone marrow, leading to their exhaustion and, ultimately, bone marrow failure.
Clinical Manifestations
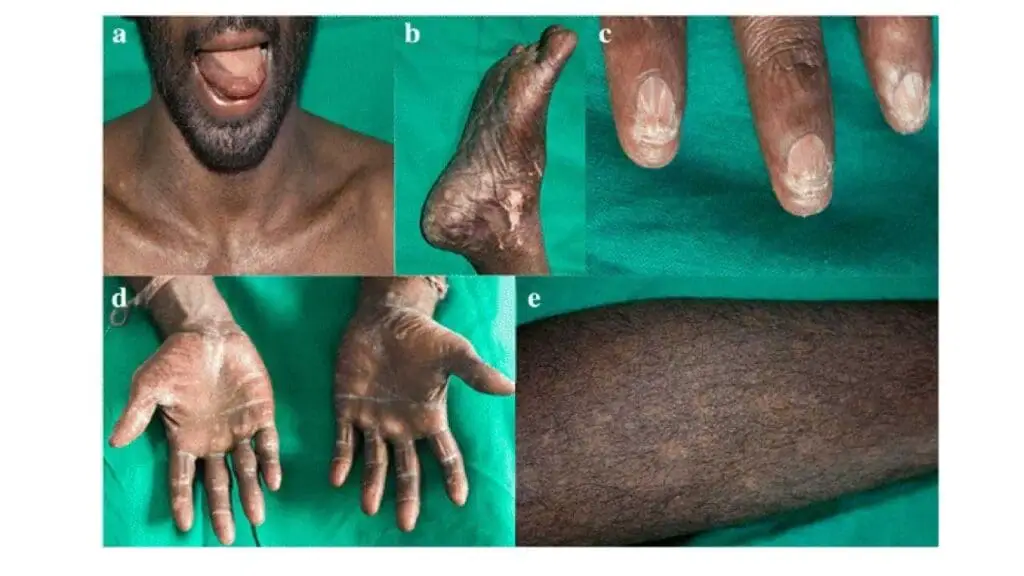
The clinical presentation of dyskeratosis congenita is highly variable, both in age of onset and severity. The classic mucocutaneous triad is a hallmark but may not be present in all individuals, especially at a young age.
- Nail Dystrophy: This is a hallmark feature in dyskeratosis congenita, affecting both fingernails and toenails. They become thin, brittle, and develop longitudinal ridges before they can eventually be lost entirely.
- Abnormal Skin Pigmentation: Dyskeratosis congenita patients develop a distinctive “lacy” or reticulated pattern of hyperpigmentation (darkening) and hypopigmentation (lightening), particularly on the neck, shoulders, and upper chest. This gives the skin a mottled appearance.
- Oral Leukoplakia: White, thickened patches appear on the mucous membranes of the mouth. This is a critical finding as these lesions have a significant potential to become cancerous (malignant transformation), specifically into squamous cell carcinoma.
Beyond these visible signs, the most life-threatening issues are the hematologic manifestations. Patients experience progressive bone marrow failure, which is the leading cause of death in this condition. This leads to pancytopenia, a severe reduction in all three major blood cell lines.
- Anemia: Low red blood cells, causing fatigue and weakness.
- Neutropenia: Low white blood cells, increasing susceptibility to infections.
- Thrombocytopenia: Low platelets, leading to easy bruising and bleeding.
Finally, while the triad and bone marrow failure are central, dyskeratosis congenita also affects multiple other organ systems, leading to a broad spectrum of complications. These include:
- Pulmonary fibrosis: Scarring of the lungs, which can lead to severe breathing problems.
- Liver disease: Cirrhosis of the liver.
- Increased risk of malignancies: This risk is a major concern. In addition to the oral leukoplakia, patients have a higher incidence of myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML), as well as solid tumors.
Laboratory Investigations and Diagnosis
The diagnostic process for dyskeratosis congenita (DC) typically begins with clinical suspicion, but it is confirmed through specific laboratory tests that investigate both the cellular and genetic abnormalities characteristic of the disease.
Clinical Suspicion
The diagnosis is first suspected when a patient presents with the classic mucocutaneous triad – the distinctive skin, nail, and oral features. The presence of additional symptoms, particularly those related to bone marrow failure or other systemic complications, further strengthens the suspicion.
Laboratory Investigations
These tests are crucial for assessing the extent of the disease and guiding further steps:
- Complete Blood Count (CBC): This is a routine but essential test. It helps identify the presence and severity of cytopenias, which are low counts of blood cells. The CBC can reveal pancytopenia, a reduction in red blood cells (anemia), white blood cells (neutropenia), and platelets (thrombocytopenia), which are hallmarks of progressive bone marrow failure.
- Bone Marrow Biopsy: When cytopenias are found, a bone marrow biopsy is often performed. This procedure allows for a microscopic examination of the bone marrow. In dyskeratosis congenita (DC), the biopsy typically shows hypocellularity, meaning there are fewer hematopoietic stem cells than expected, or even aplasia, indicating a severe loss of blood cell production.
- Telomere Length Measurement: This is the most definitive and crucial diagnostic test for dyskeratosis congenita (DC). The disease is caused by defects in telomere maintenance, leading to abnormally short telomeres. The measurement can be performed using specialized techniques:
- Quantitative PCR (qPCR): A method that compares the telomere DNA to a single-copy gene to estimate average telomere length.
- Flow Cytometry with Fluorescence In Situ Hybridization (Flow-FISH): A more advanced technique that directly measures the telomere length of individual cells, providing a more precise assessment, especially in specific cell populations like lymphocytes.
Genetic Testing
Once critically short telomeres are identified, genetic testing is used to confirm the diagnosis and determine the specific genetic cause. This involves sequencing the genes known to be associated with dyskeratosis congenita (DC), such as DKC1, TERC, and TERT. Identifying the pathogenic variant is vital for confirming the diagnosis, understanding the inheritance pattern, and providing genetic counseling to the patient and their family.
Differential Diagnosis of Dyskeratosis Congenita
| Condition | Key Distinguishing Features | How to Differentiate from DC |
| Aplastic Anemia | Bone marrow failure leading to pancytopenia. Absence of the classic mucocutaneous triad. | Absence of the characteristic skin, nail, and oral symptoms of dyskeratosis congenita. Telomere length is typically normal, unlike the abnormally short telomeres in dyskeratosis congenita. |
| Fanconi Anemia (FA) | Bone marrow failure with associated developmental abnormalities like radial ray defects (thumb and forearm malformations) and short stature. | Different spectrum of congenital abnormalities. The underlying genetic cause is a defect in DNA repair, not telomere maintenance. Diagnosis is confirmed by a chromosomal breakage test. |
| Other Telomere Biology Disorders | A group of syndromes (e.g., Hoyeraal-reidarsson syndrome) that also involve telomere defects but often present with earlier onset or more severe, multi-systemic symptoms. | Differentiated from dyskeratosis congenita by specific clinical presentation and genetic testing to identify the exact gene mutation. |
Clinical Management and Therapeutic Strategies
The treatment of dyskeratosis congenita (DC) is complex and largely supportive, aimed at managing the progressive bone marrow failure and other systemic complications. The only curative intervention for the hematologic component of the disease is a hematopoietic stem cell transplantation.
Supportive Care
Supportive care is essential for improving the quality of life and prolonging survival. It addresses the cytopenias that arise from bone marrow failure.
- Blood Transfusions: Patients with severe anemia often require regular blood transfusions to maintain adequate red blood cell counts, thereby alleviating symptoms of fatigue and preventing organ damage from tissue hypoxia.
- Hematopoietic Growth Factors: These agents stimulate the production of specific blood cell lines. Granulocyte colony-stimulating factors (G-CSFs) are used to manage severe neutropenia and reduce the risk of life-threatening infections. Similarly, thrombopoietin receptor agonists can be used to increase platelet counts and manage thrombocytopenia.
Targeted Therapy
These therapies are aimed at addressing the underlying bone marrow failure.
- Androgens: Hormones such as oxymetholone have been used to stimulate hematopoiesis and can lead to a temporary improvement in blood cell counts in some patients. However, this is not a curative treatment and is often associated with significant side effects.
- Hematopoietic Stem Cell Transplantation (HSCT): This is the only definitive curative treatment for the bone marrow failure component of dyskeratosis congenita. However, it is a high-risk procedure. Patients with dyskeratosis congenita often have pre-existing organ damage, particularly to the lungs and liver, which increases the risk of transplant-related morbidity and mortality. Therefore, the decision to pursue HSCT must be carefully considered on a case-by-case basis, and meticulous pre-transplant screening is required.
Monitoring
Given the multi-systemic nature of dyskeratosis congenita, long-term, comprehensive monitoring is critical to detect and manage complications early.
- Cancer Surveillance: Patients are at a significantly increased risk of developing various malignancies, most notably squamous cell carcinoma (SCC) of the head and neck, as well as hematologic cancers like myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML). Regular screenings for these cancers are a standard part of patient management.
- Pulmonary and Hepatic Function: Due to the risk of pulmonary fibrosis and liver cirrhosis, patients require periodic monitoring of their lung and liver function to identify and manage these complications as they arise.
Prognosis
The prognosis for individuals with dyskeratosis congenita is highly variable and depends on the specific genetic mutation and the severity of the disease’s complications. Without a successful HSCT, the leading causes of death are progressive bone marrow failure, pulmonary fibrosis, and malignancy.
Frequently Asked Questions (FAQs)
How common is Dyskeratosis congenita?
Dyskeratosis congenita is a very rare disorder, with an estimated prevalence of less than 1 in 1 million individuals. Due to its variable presentation and the fact that it may go undiagnosed, the true prevalence may be higher.
Can a person have dyskeratosis congenita without the classic triad of symptoms?
Yes. Many individuals, especially in childhood, may present with only one or two features, or with isolated bone marrow failure, making diagnosis challenging. Telomere length analysis is crucial in these cases.
Is there a cure for dyskeratosis congenita?
While hematopoietic stem cell transplantation (HSCT) can cure bone marrow failure, it does not correct the underlying genetic defect. Patients may still develop non-hematologic complications such as pulmonary fibrosis.
What is the prognosis for someone with dyskeratosis congenita?
The prognosis is highly variable and depends on the specific genetic mutation and the severity of symptoms. The median survival is around 30 years, with the most common causes of death being bone marrow failure, pulmonary fibrosis, and cancer. Early diagnosis and proactive management are key to improving outcomes.
Is Dyskeratosis congenita contagious?
No, dyskeratosis congenita is a genetic disorder and cannot be spread from person to person.
What age does dyskeratosis congenita occur?
Dyskeratosis congenita (DC) is a congenital disease, meaning it is present at birth, but the age at which symptoms appear can vary widely. This variability makes it a challenge to diagnose.
- Classic Presentation in Childhood: The classic mucocutaneous triad (abnormal skin pigmentation, nail dystrophy, and oral leukoplakia) typically begins to appear in childhood. The nail and skin changes often start before the age of 10.
- Bone Marrow Failure in Adolescence/Young Adulthood: The most life-threatening complication, bone marrow failure, usually develops later than the mucocutaneous signs. In classic DC, bone marrow failure commonly occurs by the age of 20, and in 80-90% of cases, it occurs by age 30.
- Broad Range of Onset: While the classic form presents in childhood, the overall age of diagnosis can range from infancy to late adulthood. The median age at diagnosis is around 15 years, but some individuals with milder forms may not be diagnosed until their 30s or 40s. Conversely, some severe variants of Dyskeratosis congenita (DC), like Hoyeraal-Hreidarsson syndrome, present with very early-onset symptoms in infancy.
Is dyskeratosis congenita premalignant?
Yes, dyskeratosis congenita is considered a premalignant condition. This means that individuals with dyskeratosis congenita have a significantly increased risk of developing cancer, particularly certain types of malignancies.
The increased cancer risk is one of the most serious long-term complications of the disease. The primary reason for this is the defect in telomere maintenance. Normal telomeres protect the ends of chromosomes and prevent genomic instability. In dyskeratosis congenita, the short telomeres lead to chromosomal damage and an accumulation of genetic mutations over time, which can predispose cells to malignant transformation.
Glossary of Medical Terms
- Telomere: The protective cap at the end of a chromosome that protects genetic information during cell division.
- Telomerase: An enzyme that helps to maintain telomere length by adding new DNA sequences to the ends of chromosomes.
- Bone Marrow Failure: A condition in which the bone marrow does not produce enough blood cells (red cells, white cells, and platelets).
- Pancytopenia: A condition characterized by a deficiency of all three types of blood cells (red blood cells, white blood cells, and platelets).
- Aplastic Anemia: A type of bone marrow failure in which the bone marrow stops producing new blood cells.
- Leukoplakia: A white patch or plaque that develops on the mucous membranes of the mouth and is considered a premalignant lesion.
- Pulmonary Fibrosis: A chronic lung disease in which the lungs become scarred and stiff, making breathing difficult.
- Hematopoietic Stem Cell Transplantation (HSCT): A medical procedure in which a patient’s unhealthy hematopoietic stem cells are replaced with healthy ones to restore bone marrow function.
- Reduced-Intensity Conditioning (RIC): A less toxic conditioning regimen used before HSCT to prepare the patient’s body for the transplant, often used in patients with underlying genetic predispositions to toxicity, such as DC.
Disclaimer: This article is intended for informational purposes only and is specifically targeted towards medical students. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Garofola C, Nassereddin A, Gross GP. Dyskeratosis Congenita. [Updated 2023 Jun 26]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK507710/
- Uria-Oficialdegui, M. L., Navarro, S., Murillo-Sanjuan, L., Rodriguez-Vigil, C., Benitez-Carbante, M. I., Blazquez-Goñi, C., Salinas, J. A., & Diaz-de-Heredia, C. (2023). Dyskeratosis congenita: natural history of the disease through the study of a cohort of patients diagnosed in childhood. Frontiers in pediatrics, 11, 1182476. https://doi.org/10.3389/fped.2023.1182476
- Amanda J. Walne, Laura Collopy, Shirleny Cardoso, Alicia Ellison, Vincent Plagnol, Canan Albayrak, Davut Albayrak, Sara Sebnem Kilic, Turkan Patıroglu, Haluk Akar, Keith Godfrey, Tina Carter, Makia Marafie, Ajay Vora, Mikael Sundin, Thomas Vulliamy, Hemanth Tummala, Inderjeet Dokal. Marked overlap of four genetic syndromes with dyskeratosis congenita confounds clinical diagnosis. Haematologica 2016;101(10):1180-1189; https://doi.org/10.3324/haematol.2016.147769.
- Savage S. A. (2022). Dyskeratosis congenita and telomere biology disorders. Hematology. American Society of Hematology. Education Program, 2022(1), 637–648. https://doi.org/10.1182/hematology.2022000394
- Fernández García MS, Teruya-Feldstein J. The diagnosis and treatment of dyskeratosis congenita: a review. J Blood Med. 2014;5:157-167. https://doi.org/10.2147/JBM.S47437
- Seham Khattab, Hisham Nasser, Moatasem Hussein Al-Janabi, Fouz Hasan, Dyskeratosis congenita: a rare case report, Oxford Medical Case Reports, Volume 2024, Issue 5, May 2024, omae049, https://doi.org/10.1093/omcr/omae049
- Garus, A., & Autexier, C. (2021). Dyskerin: an essential pseudouridine synthase with multifaceted roles in ribosome biogenesis, splicing, and telomere maintenance. RNA (New York, N.Y.), 27(12), 1441–1458. https://doi.org/10.1261/rna.078953.121
- Callea, M., Martinelli, D., Cammarata-Scalisi, F., Grimaldi, C., Jilani, H., Grimaldi, P., Willoughby, C. E., & Morabito, A. (2022). Multisystemic Manifestations in Rare Diseases: The Experience of Dyskeratosis Congenita. Genes, 13(3), 496. https://doi.org/10.3390/genes13030496