TL;DR
Alpha thalassemia is an inherited hemolytic anemia due deletions or point mutation (uncommon) of the α-globin gene leading to reduced production or absence of α-chains needed for hemoglobin. It has an autosomal recessive inheritance pattern.
Genetics ▾: Alpha thalassemia has an autosomal recessive inheritance pattern.
- α0-thalassemia: deletion of both α genes on the same allele (–/αα)
- α+-thalassemia: deletion of single α gene (-α/αα)
- αT-thalassemia: non-deletional mutations (αTα/αα)
Epidemiology ▾: Highest prevalence in Southeast Asia followed by West Africa, Mediterranean, Middle East, India and Pacific Islands.
Symptoms of Alpha Thalassemia ▾: The severity of symptoms in alpha thalassemia varies widely depending on the number of affected alpha-globin genes.
- Silent carrier (deletion of one gene): Usually asymptomatic.
- Alpha-thalassemia trait (deletion of two genes): No symptoms or mild fatigue.
- Hemoglobin H disease (deletion of three genes): Moderate to severe anemia, jaundice, splenomegaly, and fatigue.
- Hydrops fetalis (deletion of all four genes): Severe anemia leading to fetal death.
- Genetic Testing: To confirm the specific genetic mutation causing alpha thalassemia.
- Complete Blood Count (CBC): To assess red blood cell indices, which may show microcytosis and hypochromia.
- Peripheral Blood Smear: Hypochromic microcytic anemia with poikilocytosis.
- Brilliant Cresyl Blue (BCB): Golf-ball like inclusion bodies in the RBCs.
- Hemoglobin Electrophoresis: Presence of fast eluting bands of Hb H or Hb Barts
- HPLC: Hb H and Hb Bart’s may appear as pre-run peaks.
Treatment and Management for Alpha Thalassemia ▾:
- Mild Cases (Silent Carrier and Alpha-Thalassemia Trait): Usually asymptomatic thus no specific treatment is required.
- Moderate to Severe Cases (Hemoglobin H Disease)
- Supportive care: Includes folic acid supplementation and management of any associated symptoms.
- Blood transfusions: May be required in severe cases to treat anemia.
- Iron chelation therapy: To prevent iron overload if frequent blood transfusions are necessary.
- Splenectomy: In some cases, splenectomy may be considered to reduce hemolysis and improve symptoms.
- Hydrops Fetalis
- Prenatal diagnosis: Early detection through fetal ultrasound and blood tests.
- Intrauterine transfusion: May be performed to improve fetal outcomes.
- Abortion may be recommended.
*Click ▾ for more information
What is alpha thalassemia?
Alpha thalassemia is defined as an inherited hemolytic anemia mainly due to deletion found on the alpha globin gene(s) leading to reduced production or absence of alpha-globin chains needed for the formation of the hemoglobin tetramer.
Hemoglobinopathies is a group of genetic disorders that affect the structure or production of hemoglobin in the red blood cells. Common hemoglobinopathy disorders include:
- Sickle cell disease (SCD): A severe hemoglobinopathy caused by a single point mutation in the beta-globin gene that results in the production of abnormal hemoglobin S. Hemoglobin S molecules tend to polymerize under low-oxygen conditions, causing red blood cells to sickle or adopt a crescent-like shape. This sickling can obstruct blood flow, leading to pain, tissue damage, and a range of complications.
- Thalassemia: A group of disorders characterized by reduced or absent production of either alpha-globin (alpha-thalassemia) or beta-globin chains (beta-thalassemia), the two components of normal hemoglobin. Thalassemia can present with mild to severe symptoms, depending on the type and severity of the genetic defect.
- Hemoglobin variants: These are less common hemoglobinopathies that involve single amino acid substitutions in the alpha-globin or beta-globin chains. Some variants may have no noticeable effects, while others can cause mild symptoms or even life-threatening complications. These variants changes the structure of the hemoglobin chains making it less effective in oxygen delivery.
What is Thalassemia?
Thalassemia arises from mutations in the genes responsible for producing hemoglobin. These mutations lead to either reduced or absent production of alpha-globin or beta-globin chains, the two components of normal hemoglobin. Thus, thalassemia can be divided into alpha and beta thalassemia.
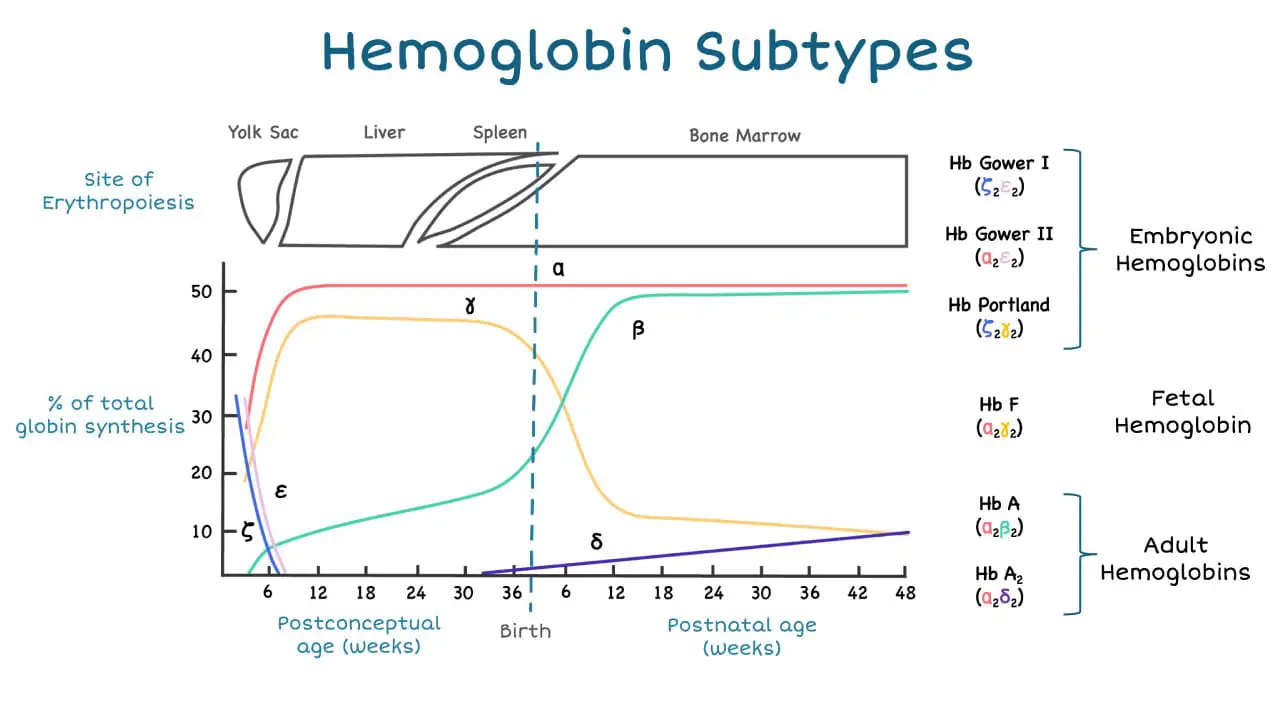
The alpha-globin gene cluster is found on chromosome 16 while the beta-globin gene cluster is found on chromosome 11. The genes in the cluster are arranged according to the order of development and at different stages of development, different hemoglobin subtypes are formed to have different oxygen affinities.
Global Prevalence and Significance of Alpha Thalassemia

Alpha thalassemia is one of the most prevalent single-gene blood disorders worldwide, affecting an estimated 270 million carriers, with the highest prevalence in Southeast Asia, the Mediterranean region, and parts of Africa and the Middle East. This widespread prevalence underscores the significant global burden of alpha thalassemia and its impact on individuals and healthcare systems worldwide.
Genetic basis of Alpha Thalassemia
There are two alpha-globin genes also known as HBA2 and HBA1 in a single allele while there is only one beta globin gene on a single allele.
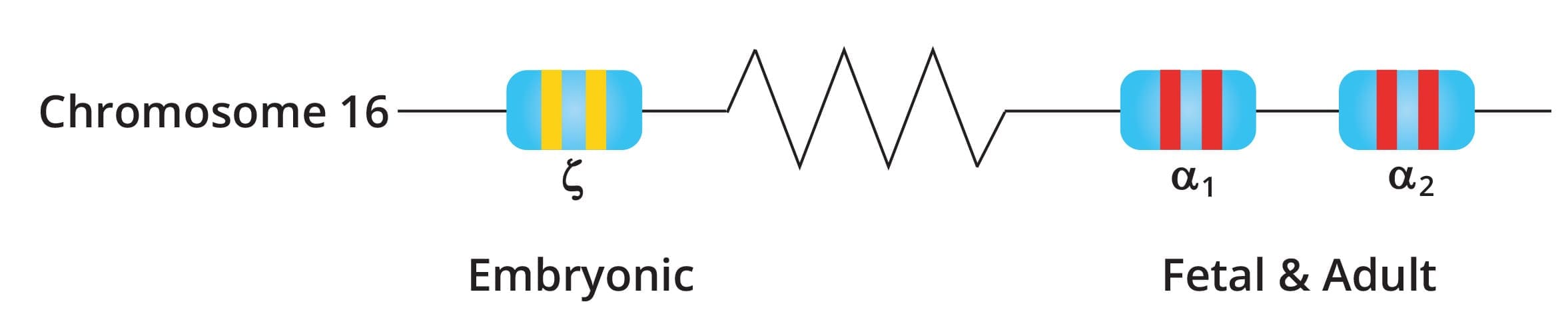
The normal α genotype is written as αα/αα. Mutations affecting α2 causes more severe anemia compared to α1 mutations as 75% of α chains are produced by α2.
When a person has alpha thalassemia, the production of the beta-globin chains is not affected while the production of the alpha-globin chain specifically is reduced or absent depending on the mutation involved.
A hemoglobin (Hb) needs 2 alpha- and 2 beta-globin chains to form a functional tetramer, thus when the alpha-globin is quantitatively reduced, there will be less Hb A available and free beta-globin chains will precipitate to form beta-globin tetramers which are also known as Hb H.
Alpha-globin expression is very high even from very young fetal age compared to beta-globin which only rises postnatally. This indicates the importance of this gene and severe mutation of this gene can cause death in utero.
Genetic Inheritance of Alpha Thalassemia
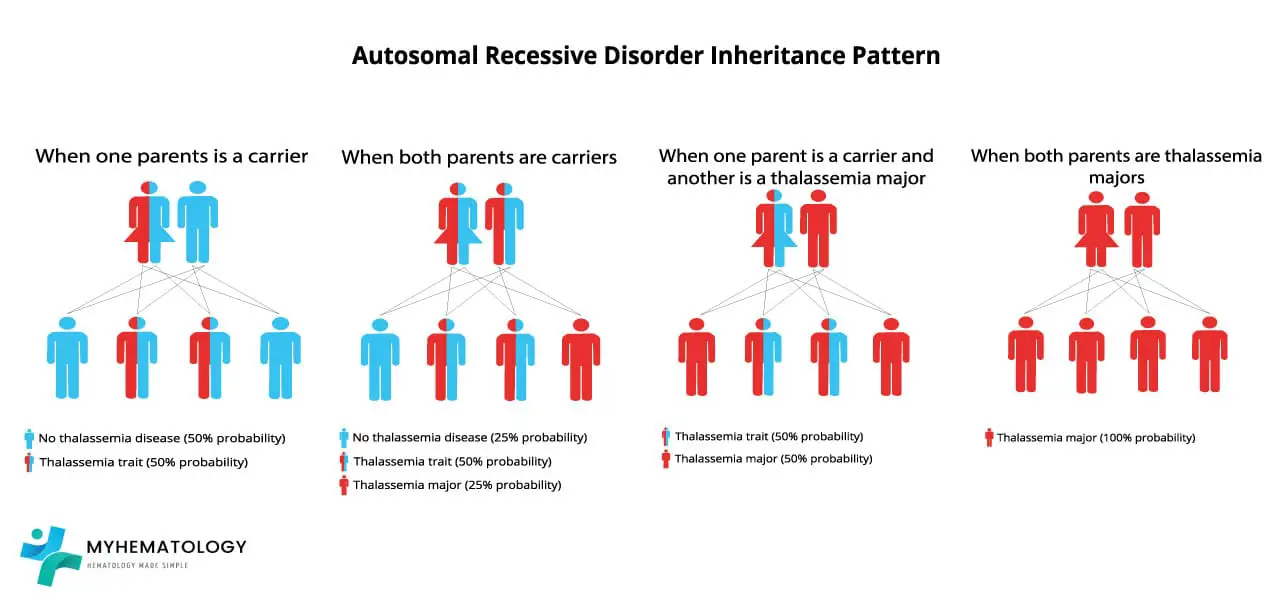

Alpha-thalassemia follows an autosomal recessive inheritance pattern. This means that the genes responsible for alpha-globin synthesis are located on non-sex chromosomes (autosomes), and both copies of the gene need to be mutated for the disorder to manifest.
To understand the inheritance pattern, let’s consider the two alleles (gene variants) of the alpha-globin gene: α and αT. The normal α allele produces functional alpha-globin chains, while the mutated αs allele produces either reduced or no alpha-globin chains.
An individual with two normal α alleles (αα) is considered a non-carrier of alpha-thalassemia and will not develop the disorder. An individual with one normal α allele and one mutated αs allele (ααT) is considered a carrier of alpha-thalassemia. Carriers typically do not experience any symptoms or health complications, but they can pass the mutated αT allele to their offspring.
The inheritance pattern becomes more apparent when considering couples with different carrier statuses.
Scenario 1: Both parents are ααT carriers
In this scenario, each parent has one normal α allele and one mutated αT allele. When they have a child, there are four possible outcomes:
- 25% chance of the child having two normal α alleles (αα) and being a non-carrier.
- 50% chance of the child having one normal α allele and one mutated αs allele (ααT) and being a carrier.
- 25% chance of the child having two mutated αs alleles (αTαT) and developing alpha-thalassemia.
Scenario 2: One parent is αα non-carrier and the other is ααT carrier
In this scenario, one parent has two normal α alleles and is not a carrier, while the other parent has one normal α allele and one mutated αs allele. When they have a child, there are two possible outcomes:
- 50% chance of the child having two normal α alleles (αα) and being a non-carrier.
- 50% chance of the child having one normal α allele and one mutated αs allele (ααT) and being a carrier.
Scenario 3: One parent is ααT carrier and the other is αTαT has alpha-thalassemia
In this scenario, one parent has one normal α allele and one mutated αT allele, while the other parent has two mutated αT alleles and has alpha-thalassemia. When they have a child, there are two possible outcomes:
- 50% chance of the child having one normal α allele and one mutated αs allele (ααT) and being a carrier.
- 50% chance of the child having two mutated αT alleles (αTαT) and developing alpha-thalassemia.
Understanding the genetic inheritance of alpha-thalassemia is crucial for genetic counseling and carrier screening. Carrier screening allows individuals to determine their risk of passing the mutated αT allele to their offspring, enabling informed decisions about family planning and preventing the transmission of alpha-thalassemia to future generations.
What are the types of mutations in alpha-thalassemia?
Alpha thalassemia mutations are mainly deletions and they usually delete one or both cis-alpha globin genes.
- Complete abolishment or deactivation of both alpha globin genes in the same allele is known as alpha0-thalassemia.
- Deletions affecting only a single gene deletion in the allele are known as alpha+-thalassemia.
- There are also non-deletional alpha+-thalassemia that are usually point mutations for example Hemoglobin Constant Spring and Hemoglobin Adana. Non-deletional alpha thalassemia has a more deleterious effect on the red blood cells as they are unstable and precipitate to cause membrane damage and release oxidative reactive species causing oxidative stress to the red cells.
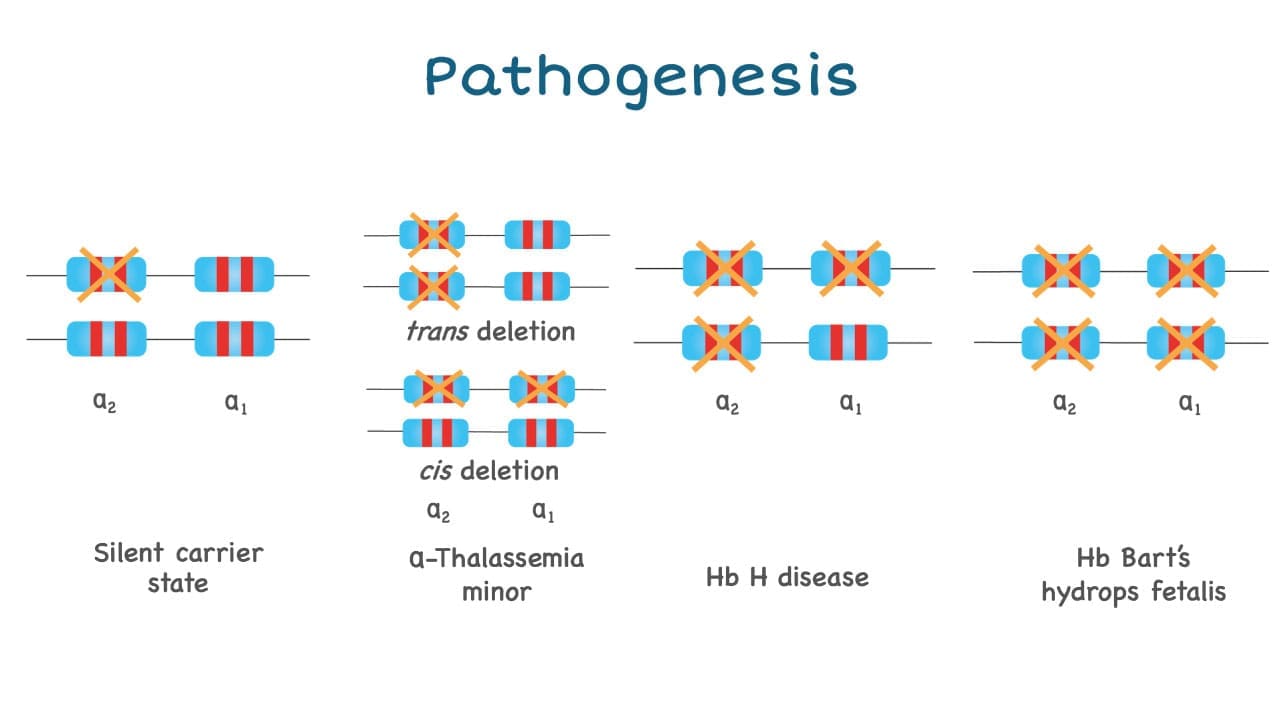
Pathogenesis of Alpha Thalassemia
The anemia caused by alpha-thalassemia is usually dependent on how many genes are deleted and as there are 4 alpha globin genes in a person.
- Deletion on only one alpha globin gene is barely noticeable and sometimes could not even be picked up by a full blood count as the values are near normal.
- Deletion of 2 genes will give rise to alpha thalassemia trait or minor and has slight microcytic hypochromic anemia picture.
- Deletion of 3 alpha globin genes leads to Hb H disease.
- Deletion of all four alpha globin genes (Hb Barts hydrops fetalis) can have a lethal effect.
Clinical Picture of Alpha Thalassemia
Alpha thalassemia presents with a spectrum of clinical manifestations ranging from mild anemia to severe life-threatening complications. The severity of the condition depends on the number of affected alpha-globin genes.
General signs and symptoms that can be observed in individuals with alpha thalassemia, particularly in more moderate to severe forms, include:
- Fatigue and Weakness: Due to the reduced oxygen delivery to tissues, individuals often experience persistent tiredness and a general feeling of weakness. This can manifest as trouble doing normal amounts of physical activity or exercise intolerance.
- Pale or Yellowish Skin (Jaundice): Anemia can cause pale skin, while the increased destruction of red blood cells can lead to a buildup of bilirubin, resulting in yellowing of the skin and the whites of the eyes (jaundice).
- Enlarged Spleen and Liver (Hepatosplenomegaly): The spleen works harder to remove damaged red blood cells, often becoming enlarged. The liver may also enlarge.
- Slow Growth and Delayed Puberty: In children with more severe forms, anemia can hinder normal growth rates and delay the onset of puberty.
- Bone Changes: The bone marrow may expand in an attempt to produce more red blood cells, leading to changes in bone structure, particularly in the face and skull. This can cause bones to widen, thin, and become brittle, increasing the risk of fractures.
- Dark Urine: The breakdown of red blood cells can lead to dark or tea-colored urine.
- Poor Appetite: Some individuals may experience a reduced appetite.
- Shortness of Breath: Reduced oxygen-carrying capacity can lead to shortness of breath, especially during physical activity.
| Type of Alpha Thalassemia | Key Symptoms |
| Silent Carrier | Typically has no symptoms. Blood tests are often normal, though red blood cells may be smaller than normal. |
| Alpha Thalassemia Trait (Minor/Carrier) | May have mild anemia. Symptoms, if present, are mild and can include mild fatigue or trouble doing normal amounts of physical activity (exercise intolerance). Red blood cells may be unusually small and pale. |
| Hemoglobin H (HbH) Disease Hb H is the tetrameric formation of 4 beta-globin chains (β4) due to the lack of alpha-globin chains and in turn lots of excess beta-globin chains. | Causes moderate to severe anemia. Symptoms include fatigue and exercise intolerance. Other symptoms can include an enlarged liver or spleen (hepatosplenomegaly) , yellowish skin (jaundice) , and leg ulcers. Symptoms may worsen with fever or exposure to certain medicines, chemicals, or infectious agents. |
| Hb Bart Syndrome (Alpha Thalassemia Major) Hb Barts is the tetrameric formation of 4 gamma-globin chains (ɣ4). Hemoglobin Barts has an extremely high affinity for oxygen, meaning it binds oxygen very tightly. This makes it difficult for the hemoglobin to release oxygen to the tissues, leading to tissue hypoxia (inadequate oxygen supply). | Causes severe anemia. Characterized by hydrops fetalis (excess fluid buildup in the body before birth). Additional signs and symptoms can include an enlarged liver and spleen , heart defects , and abnormalities of the urinary system or genitalia. In most cases, babies with this condition are stillborn or die shortly after birth. It can also cause serious complications for pregnant women, such as dangerously high blood pressure (preeclampsia), premature delivery, and abnormal bleeding. |
Laboratory Investigations
Peripheral Blood Smear
Hypochromic microcytic anaemia with some target cells can be seen in the peripheral blood smear and golf ball inclusions can be seen in brilliant cresyl blue staining (BCB).
The staining for Hb H inclusion bodies uses brilliant cresyl blue (BCB) or methylene blue (MB) as an oxidant to denature Hb H as intracellular inclusions. This method almost effectively confirms the presence of Hb H disease.
HPLC
High performance liquid chromatography (HPLC) is used to quantitatively determine the different hemoglobin subtypes available in the blood as each hemoglobin subtype has a different molecular charge and will elute at different retention times which allows them to be separated.
Hb H is a fast eluting subtype and thus will be seen as small peaks in the beginning of the run.
Hb Barts is also a fast eluting band and is easily seen as peaks in the beginning of the HPLC run too.
Capillary electrophoresis
Capillary electrophoresis is an upgraded technique to give a better resolution and separation of the hemoglobin subtypes. HPLC and capillary electrophoresis are gaining in popularity because these methods are more automated, the instruments are more user friendly, and they can be used to confirm hemoglobin variants observed with electrophoresis.
Hemoglobin electrophoresis
Hemoglobin electrophoresis is an old method which is based on the separation of hemoglobin molecules in an electric field primarily as a result of differences in total molecular charge. There are alkaline and acidic electrophoresis available.
In alkaline electrophoresis, hemoglobin molecules assume a negative charge and migrate toward the anode (positive pole). Historically, alkaline haemoglobin electrophoresis was performed on cellulose acetate medium, but it is being replaced by agarose medium. Nonetheless, because some haemoglobins have the same charge and therefore the same electrophoretic mobility patterns, hemoglobins that exhibit an abnormal electrophoretic pattern at an alkaline pH may be subjected to electrophoresis at an acid pH for definitive separation.
In an acid pH some haemoglobins assume a negative charge and migrate toward the anode, whereas others are positively charged and migrate toward the cathode (negative pole).
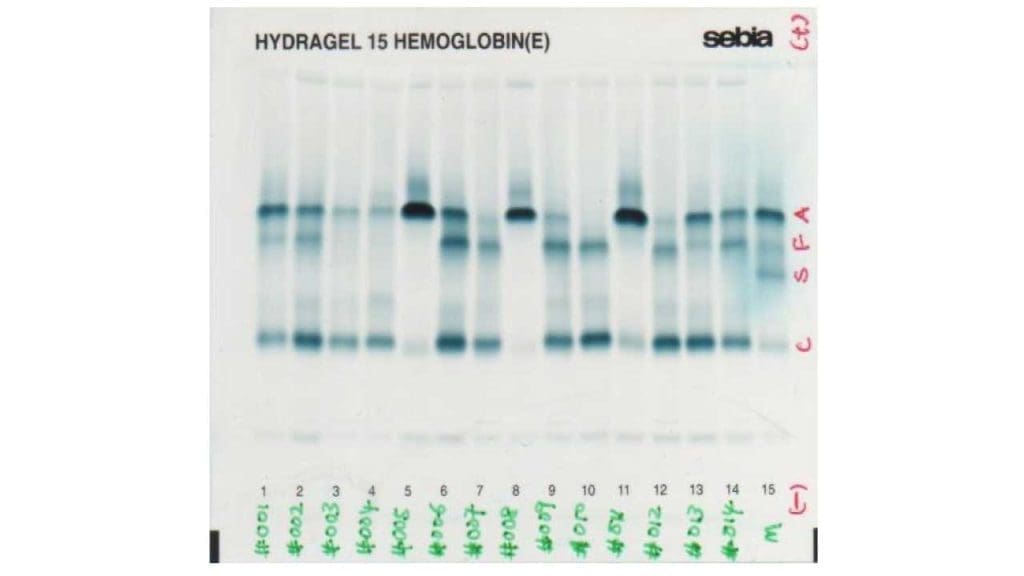
Laboratory Interpretations
Silent carriers
Silent carriers of alpha thalassemia only have one of the four genes deleted and there is a minimal decrease in the alpha-globin synthesis as it is well compensated by the other 3 functional alpha globin genes so the alpha/beta chain ratio is still quite balanced. They are usually asymptomatic and with near normal red cell indices. Through the blood count and blood film, silent carriers have minimal reduction in the red cell indices and are often undetectable in the peripheral blood film.
Alpha thalassemia trait
The alpha thalassemia trait patients have mild anemia with MCV around 65-76 fL and MCH around 22 pg. Microcytic hypochromic red cells can be seen in the blood film of alpha thalassemia trait.
Alpha thalassemia trait individuals have 2 genes deleted whether cis or trans-deletions. They have mild anemia and the microcytic and hypochromic cells are more prominent and easily detected through a full blood count and blood smear.
Hb H disease
Deletions of 3 out of the four genes will lead to Hb H disease. This compound heterozygosity leads to moderate anemia and an excess of free beta-globin chains that will precipitate to form Hb H.
Hb H will be able to be detected using the HPLC or Hb electrophoresis and the Hb H golf ball inclusions can be seen in the peripheral blood smear using the brilliant cresyl blue staining technique.
Non-deletional Hb H disease usually has a more severe picture. There is an increase in the reticulocyte count with microcytic hypochromic red cells, anisopoikilocytosis and target cells. Hb H inclusions can be seen in the blood film stained with brilliant cresyl blue stain.
Hb Barts Hydrops Fetalis
Deletion of all four alpha-globin genes is incompatible with life outside of the uterus. Absence of alpha globin chain production will lead to free gamma globin chains that will precipitate to form Hb Barts. Hb Barts has an extremely high oxygen affinity which prevents the release of oxygen to be used by the tissue thus causing tissue hypoxia and major organ failures in utero. Hb Barts hydrops fetalis babies have severe anemia and macrocytic red cells. There is severe anisopoikilocytosis with microcytic hypochromic red cells and nucleated red blood cells can be seen in the blood film. Hb Barts hydrops fetalis babies die in utero or shortly after birth if no action is taken. The confirmation of alpha globin mutations can be done through GAP PCR for deletional mutations or ARMS PCR for non-deletional mutations.
Treatment and Management of Alpha Thalassemia
| Type of Alpha Thalassemia | Treatment and Management |
| Silent Carrier (Alpha Thalassemia Minima) | No specific treatment is typically needed, as individuals usually do not experience symptoms. |
| Alpha Thalassemia Trait (Minor) | No specific treatment is usually required. Folic acid supplements may be recommended to help the body make healthy blood cells. |
| Hemoglobin H (HbH) Disease | Management often includes: – Folic acid supplements to help the body produce healthy red blood cells. – Occasional blood transfusions as needed, especially during times of infection or fever, to restore normal levels of healthy red blood cells and hemoglobin. – Iron chelation therapy to remove excess iron from the body, particularly if frequent blood transfusions are received, as iron overload can damage organs. Medications like deferasirox, deferiprone (oral), or deferoxamine (intravenous) may be used. – Splenectomy (surgical removal of the spleen) may be considered if the spleen becomes too enlarged or to reduce the need for blood transfusions. – Avoiding certain oxidant medicines is advised. – Other medicines like luspatercept (Reblozyl) may help reduce the need for blood transfusions, and hydroxyurea (Hydrea, Droxia) can lower the chances of other health problems. |
| Hb Bart Syndrome (Alpha Thalassemia Major) | This severe form usually results in stillbirth or death shortly after birth without intervention. Management options, particularly when diagnosed prenatally, may include: – Prenatal diagnosis and intrauterine transfusions to support the fetus. – Abortion may be considered as a management option. – For the rare instances when a newborn survives, lifelong blood transfusions are typically needed. – Iron chelation therapy is crucial for survivors receiving frequent transfusions to manage iron overload. – Bone marrow and stem cell transplant from a compatible donor is currently the only known cure, especially for children with severe thalassemia, potentially eliminating the need for lifelong transfusions and iron overload drugs. |
Summary of the Different Classifications
| Silent carrier state | Alpha thalassemia trait | Hb H disease | Hb Barts hydrops fetalis syndrome | |
| Pathogenesis | 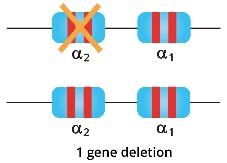 | 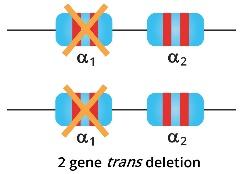 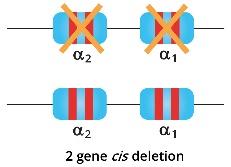 | 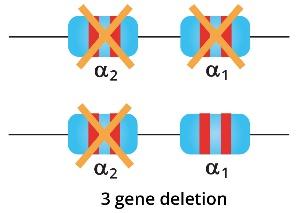 | 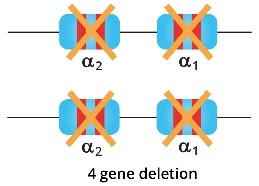 |
| Signs and Symptoms | Asymptomatic | Asymptomatic | Chronic hemolytic anemia, splenomegaly, jaundice | Severely anemic, hydropic fetus |
| Pathophysiology | α/β chain ratio is almost balanced thus no hematologic abnormalities | Low α chain production → excess unpaired β chains → Hb H (β4) → prone to oxidation → precipitation forming inclusion bodies → hemolytic anemia | No α chain production → excess unpaired γ chains → Hb Barts (γ4) → very high oxygen affinity →severely anoxic fetus | |
| Laboratory investigations | Near normal with slight reduction in MCV & MCH | Hb normal or slightly reduced. Low MCV and MCH | CBC: Anemia (Hb 7 – 11 g/dL), variable reticulocytes count (5 – 10%). PBF: Microcytic hypochromic RBCS with marked poikilocytosis including target cells. Brilliant cresyl blue stain display golf ball-like inclusion bodies in RBCs. Bone marrow: erythroid hyperplasia | PBF: Severe microcytic hypochromic anemia (Hb 3 – 8 g/dL) with numerous nucleated RBCs. Bone marrow: marked erythroid hyperplasia |
| Treatment | None required | Intermittent transfusions | Abortion as hydropic pregnancies lead to toxemia and severe postpartum hemorrhage | |
| Management | Genetic counselling and prenatal diagnosis recommended | |||
Frequently Asked Questions (FAQs)
What if both parents have alpha thalassemia trait?
There are a couple of combinations in this equation as there are 2 alpha globin genes in 1 allele so an individual will have 4 alpha globin genes.
When both parents are carriers of alpha zero (α0) thalassemia, but the chromosomes are in trans meaning in each allele, there is 1 functional alpha globin gene and 1 deleted alpha globin gene; then 100% of their offsprings will have alpha thalassemia trait.
When both parents are carriers of alpha zero thalassemia, but the deletions are in cis meaning in 1 allele both alpha genes are normal but the other allele, both alpha genes are deleted; then there is a 25% probability of their offsprings to be unaffected, 50% probability of alpha thalassemia trait offsprings and 25% probability of Hb Barts hydrops fetalis offspring.
Can a person with alpha thalassemia take iron?
Generally no, iron supplements are not recommended for people with alpha thalassemia. While they might have anemia, the underlying cause is not iron deficiency. In fact, alpha thalassemia can lead to iron overload due to ineffective red blood cell production. Taking iron supplements can worsen this and damage organs.
Is alpha thalassemia trait a disability?
Alpha thalassemia trait itself typically isn’t considered a disability. Carriers usually have no symptoms and normal health. However, there’s a rare genetic condition called Alpha-Thalassemia/Intellectual Disability Syndrome (ATRX) caused by a different gene mutation on the X chromosome. ATRX can cause intellectual disability, developmental delays, and facial features distinct from the typical alpha thalassemia trait. It’s important to differentiate between these through genetic testing.
Can an alpha thalassemia trait donate blood?
Alpha thalassemia trait carriers can potentially donate blood if their hemoglobin levels meet the minimum requirement set by the blood donation center. While the trait itself causes mild anemia, some carriers may have normal hemoglobin. It’s best to check with your local center beforehand to confirm eligibility and have your hemoglobin level tested.
What is EF Bart’s disease?
EF Bart’s disease is a rare form of thalassemia intermedia, a blood disorder characterized by moderately low red blood cell count. It arises from the co-inheritance of two abnormal genes:
- Alpha thalassemia
- Hemoglobin E (HbE): This is a variant form of hemoglobin where a single amino acid change occurs in the beta-globin chain. While HbE itself can cause a mild form of anemia, its interaction with alpha thalassemia creates a more severe condition.
Symptoms of EF Bart’s disease can include:
- Mild to moderate anemia (fatigue, weakness, pale skin)
- Splenomegaly (enlarged spleen)
- Jaundice (yellowing of the skin and eyes)
- Bone deformities (rare)
Diagnosis involves blood tests to assess red blood cell count, hemoglobin levels, and the presence of HbE and alpha-thalassemia traits. Genetic testing can confirm the specific alpha-globin gene deletions or mutations.
Treatment for EF Bart’s disease focuses on managing the symptoms and preventing complications.
Disclaimer: This article is intended for informational purposes only and is specifically targeted towards medical students. It is not intended to be a substitute for informed professional medical advice, diagnosis, or treatment. While the information presented here is derived from credible medical sources and is believed to be accurate and up-to-date, it is not guaranteed to be complete or error-free. See additional information.
References
- Higgs DR. The molecular basis of α-thalassemia. Cold Spring Harb Perspect Med. 2013 Jan 1;3(1):a011718. doi: 10.1101/cshperspect.a011718. PMID: 23284078; PMCID: PMC3530043.
- Weatherall D. 2003 William Allan Award address. The Thalassemias: the role of molecular genetics in an evolving global health problem. Am J Hum Genet. 2004 Mar;74(3):385-92. doi: 10.1086/381402. PMID: 15053011; PMCID: PMC1182250.
- Kalle Kwaifa, I., Lai, M.I. & Md Noor, S. Non-deletional alpha thalassaemia: a review. Orphanet J Rare Dis 15, 166 (2020). https://doi.org/10.1186/s13023-020-01429-1
- Songdej, D.; Fucharoen, S. Alpha-Thalassemia: Diversity of Clinical Phenotypes and Update on the Treatment. Thalass. Rep. 2022, 12, 157-172. https://doi.org/10.3390/thalassrep12040020
- Steinberg MH, Forget BG, Higgs DR, Weatherall DJ. Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management (Cambridge Medicine) 2nd Edition. 2009.
- Orkin SH, Nathan DG, Ginsburg D, Look AT, Fisher DE, Samuel Lux MD Nathan and Oski’s Hematology and Oncology of Infancy and Childhood, 2-Volume Set (Saunders) 8th Edition. 2014
- Weatherall D. Thalassaemia: The Biography (Biographies of Disease)(OUP Oxford). 2010.
- https://thalassaemia.org.cy/publications/tif-publications/guidelines-for-the-management-of-%ce%b1-thalassaemia/