Procedure-At-A-Glance
- Sample Collection: Obtain whole blood in EDTA or Heparin anticoagulated tubes.
- Cell Preparation: Isolate PBMCs using density gradient centrifugation or utilize a standard red blood cell (RBC) lysis method.
- Washing: Wash cells with flow cytometry staining buffer (PBS + BSA/FBS) and centrifuge at 250 x g.
- Viability Staining: Add live/dead viability dye and incubate in the dark.
- Surface Staining: Add fluorophore-conjugated primary antibodies targeting surface antigens and incubate.
- Fixation/Permeabilization (Optional): If targeting intracellular antigens, apply fixative, wash, and apply permeabilization buffer.
- Intracellular Staining (Optional): Add antibodies targeting intracellular markers.
- Final Wash: Wash the cells thoroughly to remove unbound antibodies.
- Acquisition: Run the sample on the flow cytometer and acquire data utilizing appropriate gating and compensation settings.
Introduction
Immunophenotyping using flow cytometry is a powerful technique used to identify and characterize cells based on the specific proteins they express on their surface or within their cytoplasm. These proteins, called antigens, act as unique markers that define a cell’s type, origin, and function. By analyzing these antigens, immunophenotyping allows scientists to:
- Differentiate between different cell types: For example, it can be used to distinguish between different types of white blood cells, such as T cells, B cells, and natural killer cells.
- Diagnose diseases: Certain diseases, such as leukemia and lymphoma, are characterized by abnormal antigen expression patterns. Immunophenotyping can help to identify these abnormal patterns and aid in diagnosis.
- Monitor treatment response: By tracking changes in antigen expression over time, immunophenotyping can be used to assess how well a patient is responding to treatment.
- Study immune function: Immunophenotyping can be used to understand how the immune system works and how it responds to different stimuli.
Immunophenotyping of blood cell surface markers using flow cytometry
Flow cytometry is a powerful and versatile technique used in various fields of biomedical research to analyze individual cells at the single-cell level. It combines the specificity of antibodies with the sensitivity of laser technology to measure multiple physical and chemical characteristics of cells suspended in a liquid stream.
Measured parameters
- Size and granularity: Based on light scattered by the cell (forward scatter and side scatter).
- Fluorescence intensity: From various markers like fluorescently labeled antibodies bound to specific cellular components.
- Internal characteristics: By using specific dyes that can enter the cell and stain specific structures.
Flow cytometry applications
- Immunophenotyping: Identifying and characterizing cell populations based on their surface and intracellular antigens.
- Cell cycle analysis: Studying cell division stages and DNA content.
- Apoptosis detection: Measuring programmed cell death.
- Calcium flux: Assessing cellular responses to stimuli.
- Protein expression analysis: Quantifying protein levels in individual cells.
Advantages
- High throughput: Analyzes thousands of cells in seconds.
- Multiparameter analysis: Measures multiple parameters simultaneously.
- Single-cell resolution: Provides information about individual cell behavior.
- Versatility: Applicable to various cell types and research questions.
Limitations
- Requires specialized equipment and expertise.
- Data analysis can be complex.
- Not suitable for studying morphological details.
Principle of Flow Cytometry
Flow cytometry works on several key principles, combining fluid dynamics, optics, and electronics to analyze individual cells.
1. Single-cell analysis: Cells are suspended in a liquid and streamed in single file through a narrow channel. The fluid flow exerts a gentle force on the cells through shear stress, ensuring they maintain a specific orientation and minimizing cell-cell interactions. This allows for individual analysis of each cell, unlike bulk methods that measure an entire population at once.
2. Laser interrogation: A laser beam shines on the cells as they flow through the channel. This laser light interacts with the cells in two main ways:
- Scattering: The cell itself and its internal structures scatter the laser light. Detectors measure the amount of light scattered forward (forward scatter, FSC) and to the side (side scatter, SSC). These measurements provide information about the cell size, shape, and granularity.
- Fluorescence excitation: If the cells are labeled with fluorescent markers (e.g., antibodies) specific to cellular components, the laser light excites these markers, causing them to emit light at different wavelengths. Detectors capture the emitted fluorescence, revealing information about the presence and abundance of specific molecules within each cell.
3. Signal detection and conversion: Light signals from scattering and fluorescence are detected by photomultiplier tubes (PMTs) and converted into electrical signals. Each PMT is designed to detect a specific wavelength of light, allowing for multiparameter analysis.
4. Data acquisition and analysis: The electrical signals are digitized and processed by a computer. Specialized software analyzes these signals, correlating them with the specific cell properties measured. This allows researchers to identify and characterize different cell populations based on their size, granularity, and specific marker expression.
Additional notes:
- Flow cytometers can have multiple lasers and fluorescent channels, enabling simultaneous measurement of various cell characteristics.
- Sorting capabilities are also available in some flow cytometers, allowing researchers to physically isolate specific cell populations based on their measured properties.
Materials
Disclaimer: This is a general protocol and may need to be adapted depending on the specific cell population of interest, antibodies used, and flow cytometer capabilities. Always consult the antibody datasheets and optimize the protocol as needed.
- Whole blood sample (anticoagulated with EDTA, Heparin or ACD)
- Flow cytometry staining buffer (PBS with BSA and FBS)
- 12 x 75 mm round-bottom polystyrene tubes
- Fluorophore-labeled primary antibodies (specific to the protein(s) of interest)
- Phosphate-buffered saline (PBS)
- Pipettes
- Ice
- Vortex mixer
- Live/dead viability dye (e.g., Annexin V-FITC)
- Permeabilization buffer (e.g., saponin)
- Blocking buffer
- Secondary antibodies (conjugated to fluorophores)
- Optional: Red blood cell (RBC) lysis buffer
- Optional: 0.45 µM cell strainer
- Optional: Non-fixable cell viability dye (impermeant nucleic acid dye)
- Optional: Fix/Lyse solution
Protocol
Sample preparation
The sample preparation step is essential for obtaining high-quality data in flow cytometry experiments.
- Isolate the cells of interest: Whole blood contains many different cell types, and you often only want to analyze specific populations. Sample preparation techniques like density gradient centrifugation or magnetic separation allow you to isolate the desired cells for targeted analysis.
- Enhance antibody binding: Antibodies bind best to exposed antigens on the cell surface. By washing cells and removing extraneous material like serum proteins, you create a cleaner environment for specific antibody-antigen interaction, improving data accuracy.
- Reduce background noise: Debris, dead cells, and other nonspecific particles can interfere with your analysis. Sample preparation techniques like centrifugation and filtration help remove these unwanted elements, reducing background noise and improving signal clarity.
- Isolate peripheral blood mononuclear cells (PBMCs) from the blood sample using density gradient centrifugation (e.g., Ficoll-Paque PLUS).
- Wash PBMCs twice with 2 mL flow cytometry staining buffer at a low speed (250 x g) for 5 minutes each time. Carefully aspirate and discard the supernatant without disturbing the pelleted cells.
- Count cells and adjust the concentration to desired volume (e.g., 1 x 106 cells/100 µL).
- Prevent cell damage by handling samples gently; this includes avoiding bubbles, vigorous vortexing, complete removal of buffer during washes, and excessive centrifugation. Gentle techniques like pipetting and low-speed centrifugation will help maintain cell integrity.
Live/dead staining
Live/dead viability staining is a critical tool for ensuring reliable and accurate analysis in flow cytometry by separating the living cells of interest from dead ones. Key functions of this step includes:
- Differentiate live from dead cells: Flow cytometry analyzes individual cells, but dead and live cells look similar based on size and shape. Live/dead dyes target specific features of dead cells, allowing you to distinguish them and focus your analysis on the healthy population.
- Accurate data interpretation: Dead cells can nonspecifically bind antibodies, altering the signal for your target marker. Excluding them ensures your data reflects the true expression levels in living cells.
- Reduce background noise: Dead cells and debris can clog instrumentation and obscure signals from live cells, making it difficult to analyze meaningful data. Live/dead staining helps filter out this background noise.
- Assess cell health and response to treatments: By measuring the live and dead cell populations, you can evaluate the overall health of your cell sample and monitor how they respond to experimental conditions or treatments.
- Optimize experimental parameters: Live/dead staining helps you optimize experimental conditions like cell culture parameters or drug concentrations by identifying their effects on cell viability.
- Add live/dead viability dye to the cell suspension according to the manufacturer’s instructions.
- Incubate for 15 minutes at room temperature (or 4°C according to the manufacturer’s instructions) in the dark.
- Wash cells twice with 2 mL flow cytometry staining buffer at a low speed (250 x g) for 5 minutes each time. Carefully aspirate and discard the supernatant without disturbing the pelleted cells.
Fixation and permeabilization (for intracellular proteins only)
In flow cytometry, fixation and permeabilization are performed for intracellular protein staining, but not for analyzing surface proteins. Proceed to the Antibody Incubation step if targeting only extracellular antigens. Key reasons for this step:
- Accessing the Inside: Cell membranes are barriers that prevent antibodies from reaching intracellular proteins.
- Fixation: This step uses chemicals (e.g., formaldehyde) to harden the cell structure, preserving the spatial arrangement of proteins within the cell.
- Permeabilization: This step uses detergents or other agents to create small pores in the cell membrane, allowing antibodies to access and bind to the intracellular proteins of interest.
- Maintaining Integrity: Without fixation, the cells would burst open due to the permeabilization process, releasing cellular contents and making analysis impossible. Fixation stabilizes the cells and prevents them from losing their shape and internal components.
- Specific Antibody Binding: Fixatives can also help reduce nonspecific antibody binding to other cellular components, improving the specificity of the staining for the target protein.
Important Notes
- Fixation can affect the antigenicity of some proteins, potentially reducing antibody binding efficiency.
- This procedure kills the cells, making them unsuitable for analyzing live cell functions.
- When both intracellular and extracellular proteins are analyzed together, cell surface staining has to be done before fixations and permeabilization.
- Centrifuge cells (250 x g for 5 minutes), discard the supernatant and resuspend in fixation buffer according to the manufacturer’s instructions.
- Incubate for a short period (e.g., 10 minutes) on ice or at room temperature (see table below).
- Wash cells twice with flow cytometry staining buffer.
- Permeabilize cells with permeabilization buffer according to the manufacturer’s instructions.
- Wash cells twice with flow cytometry staining buffer.
| Fixative Type | Incubation Time | Temperature | Suitable Proteins | Permeabilization Method |
| Formaldehyde (4%) | 10-20 minutes | 4°C | General cytoplasmic and nuclear proteins | Detergents (Triton X-100, NP-40) |
| Paraformaldehyde (4%) | 10-20 minutes | 4°C | General cytoplasmic and nuclear proteins | Detergents (Saponin, Digitonin) |
| Methanol (100%) | 5-10 minutes | -20°C | Nuclear and membrane proteins | Not required (methanol permeabilizes) |
| Acetone (100%) | 5-10 minutes | -20°C | Cytoplasmic and membrane proteins | Not required (acetone permeabilizes) |
| Glutaraldehyde (0.25%) | 15-30 minutes | 4°C | Cytoskeletal proteins | Detergents (Triton X-100) |
Additional notes
- These are general guidelines and may need to be adjusted based on your specific cell type, target protein, and desired staining intensity.
- Always consult the antibody datasheet for recommended fixation and permeabilization methods.
- Optimize fixation and permeabilization conditions through pilot experiments.
- Consider using commercially available fixation and permeabilization kits for convenience and consistency.
- Be aware of potential protein loss or antigen masking due to fixation.
Permeabilization Methods
- Detergents: Different detergents offer varying degrees of permeabilization and can affect specific protein antigenicity. Choose based on your needs.
- Enzymes: Enzymatic methods like saponin are gentler but might not be as effective for highly compact compartments.
Remember, choosing the appropriate fixation and permeabilization method is critical for successful intracellular staining. Consider your specific experiment and optimize these procedures for accurate and reliable results.
Blocking
The blocking step holds immense importance after fixation and permeabilization in flow cytometry for intracellular staining:
- Prevents non-specific antibody binding
- Fixation and permeabilization expose numerous cellular components not normally available to antibodies. Without blocking, these exposed sites can attract antibodies non-specifically, leading to:
- High background fluorescence: This obscures the true signal originating from your target protein, making data interpretation difficult.
- False positives: Nonspecifically bound antibodies can cause healthy cells to appear positive for your marker, skewing your results.
- Fixation and permeabilization expose numerous cellular components not normally available to antibodies. Without blocking, these exposed sites can attract antibodies non-specifically, leading to:
- Improves signal-to-noise ratio
- By blocking these non-specific binding sites, the blocking agent effectively “occupies” them, leaving only your target protein available for specific antibody binding. Thus,
- Reduces background fluorescence: Leading to a clearer and more accurate representation of your target protein expression.
- Increases the intensity of specific fluorescence: As antibodies are not “competing” for non-specific sites, they bind more efficiently to your target, resulting in a brighter signal.
- By blocking these non-specific binding sites, the blocking agent effectively “occupies” them, leaving only your target protein available for specific antibody binding. Thus,
- Enhances antibody specificity
- Blocking agents are often chosen based on their similarity to the secondary antibody species (e.g., using bovine serum albumin for a goat secondary antibody). This “tricks” the secondary antibody into binding preferentially to the blocking agent on non-specific sites rather than the primary antibody bound to your target protein. This further minimizes potential cross-reactivity and ensures specific signal amplification.
- Provides a stable staining environment
- Blocking buffers often contain proteins and buffers that stabilize the cell suspension and maintain optimal conditions for antibody binding. This helps prevent:
- Antibody aggregation: Leading to reduced binding efficiency and signal loss.
- Fluorescence quenching: Affecting the intensity of your signal.
- Blocking buffers often contain proteins and buffers that stabilize the cell suspension and maintain optimal conditions for antibody binding. This helps prevent:
- Centrifuge cells (250 x g for 5 minutes), discard the supernatant and resuspend in blocking buffer according to the manufacturer’s instructions.
- Incubate for 15-30 minutes on ice or at room temperature to block nonspecific antibody binding.
- Wash cells twice with flow cytometry staining buffer.
Antibody incubation
The antibody incubation step serves two main purposes:
- Specific binding to target molecules: Primary antibodies are specifically designed to recognize and bind to unique epitopes (protein structures) on the surface or inside the cell. During the incubation step, primary antibodies interact with the cell suspension and attach to these specific target molecules.
- Signal amplification (indirect staining): In most cases, primary antibodies themselves don’t have any fluorescent or detectable tags. To convert the specific binding event into a measurable signal, secondary antibodies conjugated to fluorescent dyes are used. In the subsequent incubation step, these secondary antibodies specifically recognize the constant region (Fc fragment) of the primary antibody attached to the target molecule. This creates a “bridge” between the target and the fluorescent dye, amplifying the signal for detection by the flow cytometer.
Important factors for optimized antibody incubation
- Antibody concentration: Use the optimal concentration of each antibody (primary and secondary) to achieve specific binding without saturating the target or generating excessive background signal.
- Incubation time and temperature: Follow the manufacturer’s recommendations for each antibody, considering parameters like antigen expression level, cell type, and desired staining intensity.
- Buffer composition: Use an appropriate buffer containing stabilizing agents (e.g., BSA) to minimize non-specific interactions and maintain cell viability.
- Wash steps: Following each incubation, washing steps are crucial to remove unbound antibodies and reduce background noise.
Additional notes
In some rare cases, direct-conjugated (fluorescently labeled) primary antibodies are used, eliminating the need for secondary antibodies and simplifying the process. Specific incubation details will vary depending on the chosen staining protocol, target antigen, and cell type. Always refer to specific antibody datasheets and optimize protocols for your experiment.
Direct antibody labeling
- Add the primary antibody directly conjugated to a fluorophore to the cell suspension at the recommended concentration.
- Incubate for 30-60 minutes on ice or at room temperature in the dark.
- Wash cells twice with flow cytometry staining buffer.
Indirect antibody labeling
- Add the unconjugated primary antibody to the cell suspension at the recommended concentration.
- Incubate for 30-60 minutes on ice or at room temperature.
- Wash cells twice with flow cytometry staining buffer.
- Add the secondary antibody conjugated to a fluorophore at the recommended concentration.
- Incubate for 30 minutes on ice or at room temperature in the dark.
- Wash cells twice with flow cytometry staining buffer.
Additional notes
The same principles can be applied to perform multicolor flow cytometry, utilizing multiple sets of conjugated antibodies to simultaneously analyze various targets. However, choosing fluorophores with minimal emission spectrum overlap is crucial to accurately distinguish and measure signals from each antibody population.
Detection and data analysis
- Resuspend cells in flow cytometry staining buffer and acquire data using the flow cytometer.
- Collect data on relevant parameters, including forward scatter (FSC), side scatter (SSC), and specific fluorescence channels corresponding to the chosen dyes.
- Analyze the data using flow cytometry software to identify and characterize cell populations based on their expression of the protein of interest and viability staining.
Additional notes
- Isolate specific cell populations using surface markers through additional antibody labeling before proceeding with intracellular staining.
- Optimize antibody concentrations and incubation times based on the manufacturer’s recommendations and your specific experiment.
- Include appropriate controls such as unstained cells and cells stained with secondary antibody only to assess background fluorescence.
- Use proper gating techniques to define and analyze specific cell populations based on their marker expression.
Intracellular and Extracellular Protein Detection
If you need to stain for both intracellular and extracellular protein detection in flow cytometry, the procedure differs slightly from the typical approach.
1. Staining: Follow your usual staining protocol for extracellular proteins. This includes blocking, primary antibody incubation, washes, and (if applicable) secondary antibody incubation and washes.
2. Permeabilization: After staining for extracellular proteins, without washing off the bound antibodies, proceed with permeabilization. Choose a method suitable for your target intracellular antigen (e.g., detergent-based or enzymatic). The choice of permeabilization agent might depend on the properties of your extracellular antibody and may require optimization to balance accessibility of both markers.
3. Intracellular staining: Once permeabilized, stain your cells for the intracellular protein. This includes:
- Blocking (if needed, depending on the permeabilization method).
- Incubation with the primary antibody specific to the intracellular protein.
- Washes to remove unbound primary antibody.
- Secondary antibody incubation and washes (if using an indirect staining method).
4. Fixation: Finally, fix the cells. This step preserves the binding of both extracellular and intracellular antibodies and stabilizes the cells for further analysis. Choose a fixation method compatible with your chosen permeabilization and antibody pairs. Remember, some fixatives can affect antigenicity, so optimization might be required.
5. Analysis: Acquire data on your flow cytometer using appropriate excitation and emission filters to capture signals from both the extracellular and intracellular markers.
Important notes
- This approach requires careful optimization to balance accessibility of both markers and minimize unwanted interactions.
- Consult specific datasheets for both extracellular and intracellular antibodies to ensure compatibility with the chosen fixation and permeabilization methods.
- Consider using commercially available fixation/permeabilization kits specifically designed for intracellular staining after surface staining.
- Be aware of potential signal quenching or masking of antigens due to fixation.
- Perform control experiments to assess background staining and specificity of both markers.
This protocol provides a general framework. Remember to consult specific datasheets and adapt the protocol accordingly for your specific needs and target protein.
Interpretation
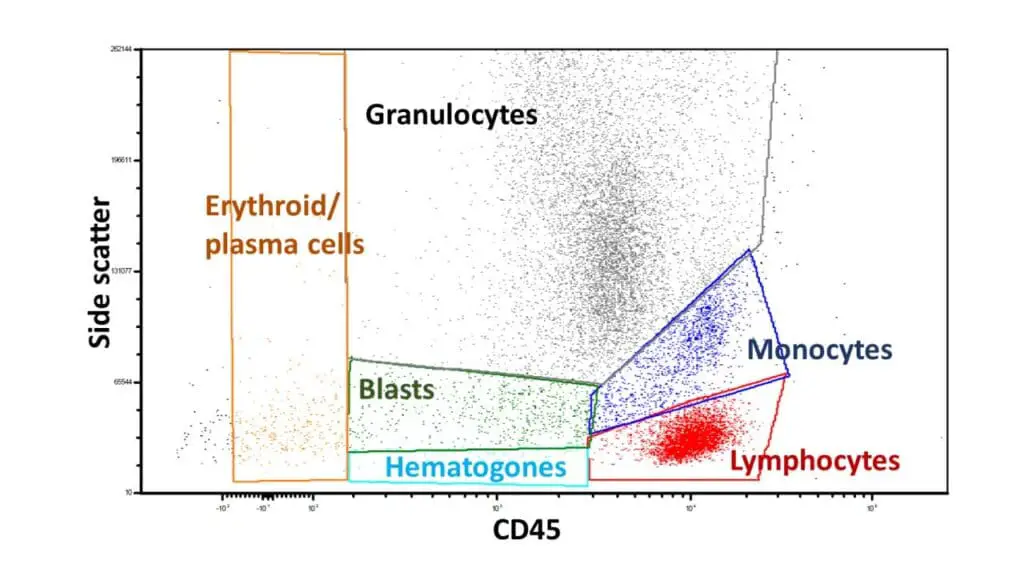
Gating Strategies
CD45 vs. Side Scatter (SSC) Gating
This is the gold standard for analyzing peripheral blood and bone marrow. Instead of relying solely on size (FSC), we use the intensity of the leukocyte common antigen (CD45).
- Lymphocytes: High CD45 expression and very low SSC (small, non-granular).
- Monocytes: High CD45 and medium SSC (larger with some internal complexity).
- Granulocytes: Lower CD45 expression but very high SSC (highly granular).
- Blasts: Typically “CD45 dim” and low SSC. This “blast window” is crucial for identifying acute leukemias that might otherwise be hidden among normal lymphocytes.
- Erythroid precursors: High side scatter, variable CD45
- Plasma cell: Variable side scatter, moderate CD45
- Hematopoietic progenitor cells: Low side scatter, low CD45
Sequential Gating
To identify a specific subset (e.g., T-helper cells), a hierarchy is followed:
- Singlet Gate: FSC-H vs FSC-A to exclude cell clumps or doublets.
- Viability Gate: Excludes cells that have taken up a live/dead dye.
- CD3+ Gate: Selects all T-lymphocytes.
- CD4+ Gate: Final selection of T-helper cells within the CD3+ population.
Core Standardized Panels Commonly Utilized in Clinical Laboratories
| Clinical Indication / Panel Type | Key Markers (Surface & Intracellular) | Target Cell Population | Primary Diagnostic Utility |
| Acute Leukemia Triage (Lineage Assignment) | Immaturity: CD34, CD45 (dim), HLA-DR Myeloid: CD117, CD13, CD33, cMPO B-Lymphoid: CD19, cCD79a, cCD22 T-Lymphoid: cCD3, CD7 | Blasts (immature hematopoietic precursors) | Differentiating Acute Myeloid Leukemia (AML) from B-cell or T-cell Acute Lymphoblastic Leukemia (ALL) and identifying mixed-phenotype acute leukemias. |
| B-Cell Chronic Lymphoproliferative Disorders (B-CLPD) | Pan-B: CD19, CD20 Subtyping: CD5, CD10, CD23, CD200, FMC7, CD103 Clonality: Surface Kappa (sκ) & Lambda (sλ) light chains | Mature B-lymphocytes | Diagnosing and subtyping mature B-cell neoplasms (e.g., distinguishing Chronic Lymphocytic Leukemia from Mantle Cell Lymphoma or Follicular Lymphoma based on CD5/CD10/CD23 expression). |
| T-Cell and NK-Cell Neoplasms | Pan-T/Subset: CD2, CD3, CD4, CD5, CD7, CD8 NK/Other: CD56, CD16, CD57, TCR-alpha/beta, TCR-gamma/delta | Mature T-lymphocytes and Natural Killer (NK) cells | Identifying aberrant loss of pan-T markers or skewed CD4:CD8 ratios in conditions like T-cell large granular lymphocytic leukemia or Sézary syndrome. |
| Plasma Cell Dyscrasias | Identification: CD38, CD138, CD45 (often negative/dim) Aberrancy: CD56, CD117, CD19, CD27, CD81 Clonality: Intracellular Kappa (cκ) & Lambda (cλ) | Plasma cells | Detecting monoclonal, aberrant plasma cell populations in Multiple Myeloma, Smoldering Myeloma, and Monoclonal Gammopathy of Undetermined Significance (MGUS). |
| Paroxysmal Nocturnal Hemoglobinuria (PNH) Screening | WBC: CD15, CD24, CD14, CD59, FLAER (Fluorescent Aerolysin) RBC: CD59, CD235a | Neutrophils, Monocytes, Erythrocytes | Detecting the absence or deficiency of GPI-anchored proteins, which definitively confirms the presence and size of PNH clones |
| Myelodysplastic Syndromes (MDS) Assessment | Myeloid/Monocytic: CD11b, CD13, CD14, CD16, CD33, CD64, HLA-DR Erythroid: CD71, CD105 | Maturing granulocytes, monocytes, and erythroid precursors | Evaluating aberrant maturation patterns, abnormal antigen expression intensity, and lineage infidelity to support an MDS diagnosis |
Frequently Asked Questions (FAQs)
How long can peripheral blood samples be stored before flow cytometry analysis?
Ideally, blood samples should be analyzed within 24 to 48 hours of collection. Viability and the expression of certain surface markers degrade over time. Samples should be kept at room temperature and never frozen prior to whole-blood processing or PBMC isolation.
Why is CD45 vs. Side Scatter gating preferred in hematology?
CD45 is the leukocyte common antigen. Plotting CD45 expression against Side Scatter (granularity) allows for the precise separation of distinct white blood cell populations. Blasts typically exhibit dim CD45 expression and low side scatter, making this gating strategy essential for identifying leukemic cell populations accurately.
What is the purpose of fluorescence compensation?
When using multiple fluorescent antibodies in a single tube, the light emitted by one fluorochrome can easily bleed into the optical detector meant for another. Compensation subtracts this overlapping signal, ensuring that the detected fluorescence in a specific channel is accurately attributed to the correct marker.
What are the useful tips for choosing suitable fixation and permeabilization methods for intracellular staining?
Consider your target protein
- Location: Does the protein reside in a specific cellular compartment (e.g., nucleus, mitochondria)? Different fixatives and permeabilization agents have varying abilities to access different compartments.
- Sensitivity: Is the protein sensitive to harsh fixation or permeabilization conditions? Some antigens might require milder methods to preserve their binding sites for antibodies.
Evaluate the fixation method
- Fixative type: Consider using formaldehyde-based fixatives for general protein preservation or methanol-based fixatives for better membrane preservation depending on your needs.
- Fixation duration and temperature: Optimize the duration and temperature of fixation to balance protein preservation with antigen accessibility. Short, cold fixes often maintain better antigenicity.
Choose the permeabilization method
- Method type: Choose between detergent-based (e.g., Triton X-100) or enzymatic (e.g., saponin) methods based on the level of permeabilization required and potential impact on your target protein.
- Permeabilization duration and concentration: Optimize the duration and concentration of the permeabilization agent to achieve adequate access to intracellular antigens while minimizing non-specific binding.
Additional tips
- Perform pilot experiments: Test different fixation and permeabilization conditions on a small subset of your cells to find the optimal balance for your protein of interest.
- Use positive and negative controls: Include unstained cells and cells stained with only secondary antibody to assess background staining and antibody specificity.
- Optimize antibody concentration: Use the lowest effective concentration of antibody to minimize non-specific binding and improve signal-to-noise ratio.
- Consult datasheets and literature: Refer to antibody datasheets and scientific literature for recommendations on compatible fixation and permeabilization methods for your specific target protein.
- Seek expert advice: If unsure about the best approach, consult your flow cytometry facility or an experienced researcher for guidance.
What are the applications for immunophenotyping by flow cytometry?
Immunophenotyping by flow cytometry finds diverse applications across various medical and research fields, largely due to its ability to identify and characterize specific cell populations based on their surface and intracellular protein expression. Here are some key examples:
1. Diagnosis and classification of blood cancers
Immunophenotyping plays a crucial role in diagnosing and classifying diverse blood cancers like leukemia, lymphoma, and multiple myeloma. By analyzing the expression patterns of specific cell surface markers on blood cells, doctors can distinguish between different types of cancer and determine the stage of the disease. This information is critical for making informed treatment decisions.
2. Monitoring treatment response and prognosis
After diagnosing a blood cancer, immunophenotyping can be used to monitor the patient’s response to treatment. By tracking changes in the expression of specific markers on cancer cells over time, doctors can assess the effectiveness of therapy and adjust it as needed. Additionally, immunophenotyping can help predict the patient’s prognosis and risk of recurrence.
3. Evaluating immune system function
Flow cytometry immunophenotyping plays a valuable role in assessing the immune system’s health and function. By analyzing the number and types of different immune cells, as well as their activation state, clinicians can diagnose immune deficiencies, autoimmune diseases, and allergies. This information helps guide further investigations and personalized treatment strategies.
4. Studying cell activation and differentiation
Researchers utilize immunophenotyping to study how cells activate and differentiate into different types during development, immune response, and wound healing. By analyzing marker expression changes during these processes, they gain valuable insights into cellular signaling pathways and regulatory mechanisms.
5. Investigating autoimmune diseases
In autoimmune disorders, the immune system mistakenly attacks healthy tissues. Immunophenotyping helps identify these aberrant immune cell populations and analyze their activation state, providing crucial information for understanding the disease pathogenesis and developing targeted therapies.
6. Transplant compatibility testing (Flow cytometry crossmatch)
Before organ transplantation, immunophenotyping is used to analyze the recipient’s immune cells and assess their compatibility with the donor organ. This helps minimize the risk of transplant rejection and improve patient outcomes.
7. Studying infectious diseases
Flow cytometry immunophenotyping can be used to identify and characterize pathogens (like viruses and bacteria) based on their unique surface protein expression patterns. This information is valuable for diagnosing infections, assessing their severity, and developing new therapeutics.
8. Drug discovery and development
Researchers use immunophenotyping to evaluate the effects of new drugs on specific cell populations, both in vitro and in vivo. This allows them to assess the drug’s efficacy and potential side effects, aiding in the development of safe and effective therapies.
Overall, immunophenotyping by flow cytometry offers a powerful tool for both clinical and research applications, providing valuable insights into cellular characteristics, function, and abnormalities across various disciplines.
What are the methods of immunophenotyping?
While flow cytometry is the most widely used and versatile method for immunophenotyping, other techniques also hold merit depending on your specific needs:
Flow cytometry
The gold standard for immunophenotyping due to its ability to analyze large numbers of cells simultaneously while capturing multiple parameters (surface and intracellular proteins). The rapid and high-throughput analysis enables detailed characterization of cell populations based on marker expression. Limitations include requiring specialized equipment and expertise, potential challenges with live cell analysis, and susceptibility to staining artifacts.
Immunohistochemistry
It utilizes antibodies and chromogenic or fluorescent tags to visualize protein expression within tissue sections. It offers spatial context for marker expression within tissues, allowing analysis of cell localization and interactions. Limited to analyzing small numbers of cells and provides qualitative rather than quantitative data.
Immunocytochemistry
Similar to immunohistochemistry but performed on isolated cells, providing insights into individual cell marker expression. It offers detailed cellular morphology information alongside protein expression analysis. Limited to analyzing small numbers of cells and requires careful cell preparation techniques.
Western blotting
It detects and quantifies protein expression levels in whole cell lysates. It is useful for validating antibody specificity and analyzing protein modifications or signaling pathways but does not provide information about individual cell characteristics or cellular distribution of proteins.
Enzyme-linked immunosorbent assay (ELISA)
It is primarily used for quantifying soluble proteins in various biological samples. It can be adapted to detect cell surface markers indirectly by capturing them from cell lysates or supernatants. It offers high sensitivity but limited information about specific cell populations or marker distribution.
Emerging methods
- Mass cytometry: Offers ultra-high-dimensional analysis of multiple markers simultaneously with superior sensitivity compared to flow cytometry.
- Imaging flow cytometry: Combines flow cytometry with high-resolution imaging, providing both quantitative data and morphological information on individual cells.
- Microfluidic technologies: Enable automated and miniaturized immunophenotyping workflows, potentially increasing accessibility and portability.
What is immunophenotyping of whole blood?
Immunophenotyping of whole blood, sometimes referred to as leukocyte immunophenotyping or whole blood immunoassay, analyzes the individual cells within your entire blood sample without prior separation of specific cell types. This differs from standard immunophenotyping, which typically isolates white blood cells first.
Can flow cytometry detect leukemia or lymphoma?
Yes, flow cytometry is a crucial tool for detecting and diagnosing leukemia and lymphoma, playing a vital role in the process alongside other tests and clinical information.
1. Identifying abnormal cell populations
Flow cytometry utilizes specific antibodies targeting unique markers on the surface and sometimes inside blood cells.
- In leukemia, it detects the presence of abnormal white blood cells (blasts) with characteristic marker patterns not found in healthy cells.
- In lymphoma, it detects the presence of abnormal lymphoid cells with distinctive marker patterns different from healthy counterparts.
2. Classifying leukemia or lymphoma type
Different leukemias express distinct marker profiles. By analyzing these patterns, flow cytometry helps classify leukemia type, including acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), chronic lymphocytic leukemia (CLL), and chronic myeloid leukemia (CML). This classification is critical for choosing the most suitable treatment approach, as different types require specific therapies.
Different lymphomas express specific marker profiles. By analyzing these patterns, flow cytometry helps classify lymphoma type, including Hodgkin lymphoma (HL) and non-Hodgkin lymphoma (NHL) further subdivided into multiple subtypes. This classification is critical for choosing the most suitable treatment approach, as different types require specific therapies.
3. Determining minimal residual disease (MRD)
After treatment, even small numbers of leukemic or lymphoma cells can remain, known as MRD. Flow cytometry, with its high sensitivity, can detect these rare populations, aiding in assessing treatment effectiveness and predicting potential relapse risk. Identifying MRD allows for early intervention or adjustments to treatment plans, improving patient outcomes.
4. Monitoring treatment response
By tracking changes in blast or abnormal cell numbers and marker expression over time throughout treatment, flow cytometry helps monitor the patient’s response to therapy. A decrease in these cells and altered marker patterns indicate a positive response, while persistent abnormal cells might suggest treatment resistance or relapse.
5. Identifying specific genetic abnormalities
Certain leukemias or lymphomas are associated with specific genetic mutations. Flow cytometry can be combined with advanced techniques like fluorescence in situ hybridization (FISH) to directly visualize these mutations within individual cells, aiding in diagnosis and targeted therapy selection.
**Flow cytometry isn’t the only tool for leukemia and lymphoma diagnosis, but it plays a central role:
- Offering rapid and detailed characterization of individual cells.
- Providing precise differentiation between normal and abnormal populations.
- Contributing to accurate leukemia classification and guiding treatment decisions.
- Monitoring treatment response and identifying MRD for improved patient care.
Can flow cytometry detect antibodies?
Flow cytometry itself does not directly detect antibodies, but it can be used in conjunction with specific tools and techniques to achieve this purpose.
1. Indirect detection of antibodies using antigen-coupled beads
In this approach, beads are coated with antigens specific to the antibody of interest. When the serum sample containing the antibody is incubated with these beads, the antibody binds to its corresponding antigen on the bead surface. Next, fluorescently labeled secondary antibodies specifically targeting the Fc region of the bound antibody are added. These secondary antibodies then bind to the primary antibodies, generating a fluorescent signal on the bead. The flow cytometer detects the fluorescent signal intensity on each bead, which indirectly reflects the amount of specific antibody present in the original sample.
2. Flow-based immunoassays
These assays utilize immobilized antigens on a microfluidic chip within the flow cytometer. The sample containing the antibody of interest is passed through the chip, allowing the antibody to bind to the immobilized antigen. Similar to the beads approach, fluorescently labeled secondary antibodies are then used to detect bound primary antibodies, generating a signal proportional to the initial antibody concentration.
3. Detection of antibody-producing cells
Flow cytometry can be used to identify individual cells within a population that are actively producing specific antibodies. This is achieved by staining the cells with fluorescently labeled antibodies targeting intracellular components involved in antibody production (e.g., B cell activating factor). By analyzing the expression pattern of these markers alongside other cell surface markers, researchers can identify and quantify antibody-producing cells within a complex sample.
Limitations of flow cytometry for antibody detection
- Requires specialized reagents and expertise in designing and optimizing the assay.
- Sensitivity might be lower compared to established techniques like ELISA.
- Not suitable for detecting all types of antibodies or assessing their functional properties.
How accurate is flow cytometry?
Flow cytometry accuracy depends on various factors, making it a broad spectrum with diverse considerations.
Overall Accuracy
- High precision: Repeated measurements on identical samples usually show consistent results, indicating high reproducibility within a specific setting.
- Variable sensitivity: Depends on the specific application, desired cell population, and available technology. It can range from very high (detecting rare cell populations) to moderate (identifying larger cell subsets).
- Specificity: Varies based on the chosen antibodies and their ability to uniquely target the intended cell population without binding to unintended cells. High specificity is crucial for accurate identification and minimal background noise.
Factors Affecting Accuracy
- Instrument calibration and maintenance: Regular calibration and proper maintenance ensure the flow cytometer functions optimally, influencing sensitivity, resolution, and reproducibility.
- Sample preparation and staining: Careful cell isolation, proper antibody concentrations, and controlled incubation times are crucial for accurate marker detection and population identification.
- Data analysis and interpretation: Expertise in software usage and understanding of marker expression patterns are essential for drawing accurate conclusions from the generated data.
- Choice of antibodies: Highly specific and validated antibodies targeting relevant markers are paramount for precise identification and minimal cross-reactivity.
Specific Examples
- Immunophenotyping for leukemia: Accuracy can be very high due to well-established marker panels and experienced personnel, aiding precise diagnosis and classification.
- Detecting rare cell populations: Sensitivity becomes particularly important, and specialized techniques might be needed to achieve the accuracy required for research or diagnosis.
- Quantifying marker expression levels: Standardization and careful data analysis are crucial for accurate quantification, which might be less precise than simple cell population identification.
Limitations
- Interpretation subjectivity: Requires expertise in analyzing marker expression patterns and considering clinical context for accurate interpretation.
- Technical limitations: Sensitivity might be limited for very rare cell populations, and instrument-related factors can influence accuracy.
- Not a standalone diagnostic tool: Flow cytometry findings need integration with other clinical information and tests for a comprehensive diagnosis.
Can flow cytometry detect metastasis?
The ability of flow cytometry to detect metastasis depends on several factors:
Direct detection
Generally, flow cytometry cannot directly detect metastasis within tissues or organs. This is because flow cytometry typically analyzes individual cells in suspension, while metastasis involves cancer cells adhering and growing within tissues.
However, in some specific cases, flow cytometry can be used to analyze circulating tumor cells (CTCs) in the blood. These are cancer cells that have shed from the primary tumor and entered the bloodstream, potentially leading to metastasis. Flow cytometry can detect and characterize CTCs based on their size, surface markers, and even intracellular features. However, CTCs are rare, and detecting them requires sensitive techniques and specialized protocols, making it not a routine tool for metastasis detection.
Indirect detection
Flow cytometry can be used indirectly to assess the risk of metastasis or monitor patients for potential recurrence. This can be done by:
- Analyzing immune system function: Evaluating the number and function of different immune cell types like lymphocytes and natural killer cells can provide insights into the body’s ability to fight cancer and control its spread.
- Detecting changes in marker expression: Monitoring changes in the expression of specific markers on tumor cells isolated from biopsies or other sources can potentially indicate their aggressive behavior and metastatic potential.
Limitations
- Flow cytometry cannot definitively diagnose metastasis without additional information from imaging or biopsy results.
- Detecting CTCs remains a research area with ongoing advances in sensitivity and specificity needed for wider clinical application.
Does flow cytometry damage cells?
Flow cytometry can certainly damage cells to some extent, but the degree of damage depends on various factors.
Sample preparation
- Concentration: Using cell concentrations within the recommended range helps minimize stress and potential damage. Overcrowding cells can lead to physical interactions and stress.
- Temperature: Maintaining appropriate temperatures during processing and analysis is crucial. Cold temperatures slow down cellular processes and reduce damage, while excessively high temperatures can be detrimental.
- Buffers and enzymes: Choosing buffers with appropriate pH and osmolarity minimizes stress on cells. Harsh enzymes used in cell isolation or staining can also cause damage.
Flow cytometer settings
- Laser power: Lasers are necessary for exciting fluorescent dyes, but using higher power than needed can induce phototoxicity, damaging cellular components.
- Fluidics pressure: Too much pressure while forcing cells through the flow cytometer can cause physical stress and membrane disruption.
- Flow rate: Slowing down the flow rate allows for gentler analysis and reduces shear stress on cells.
Overall impact
- Mild to moderate damage: In most cases, flow cytometry causes only mild to moderate damage, not compromising the ability to extract reliable information for intended purposes. This is especially true for short analysis times and optimized instrument settings.
- Cell viability assays: When assessing cell viability with flow cytometry, specific stains are used to differentiate live and dead cells. While the staining procedures themselves might cause some damage, they offer valuable insights into cell health and response to various stimuli.
- Cell sorting: Cells separated using flow cytometers for further analysis or culture might experience higher stress due to increased pressure and sorting mechanisms. However, optimized protocols and gentle collection methods can minimize damage and maintain cell viability.
Alternatives and considerations
- Alternatives: Other techniques like microscopy or microfluidics may offer gentler analysis depending on the specific needs and cell types.
- Balancing information and damage: Carefully weighing the information gained from flow cytometry against the potential cell damage is crucial. Optimizing protocols and settings reduces damage while ensuring reliable data acquisition.
Does flow cytometry count dead cells?
Whether flow cytometry can count dead cells depends on how “dead” the cells are and the techniques used. Generally, flow cytometry cannot directly count truly dead cells that have lost membrane integrity and leaked their contents.
Reasons why
- Membrane impermeable dyes: Most dyes used in flow cytometry don’t pass through healthy cell membranes. Thus, they can’t stain the internal components of truly dead cells, making them invisible to the instrument.
- Loss of cellular structure: Dead cells often lose their shape and volume, making them difficult to identify based on size and scatter properties measured by flow cytometry.
However, there are strategies to estimate or exclude dead cells
- Viability dyes: Specific dyes like propidium iodide (PI) or DAPI stain the nucleic acids of dead cells after their membranes become compromised. This allows estimating the proportion of dead cells in a population.
- Exclusion dyes: Dyes like Trypan blue are impermeable to healthy cells but enter dead cells with compromised membranes. By comparing the number of stained and unstained cells, you can estimate the dead cell population.
- Gating strategies: By analyzing scatter properties (forward and side scatter) and fluorescence intensity of viable dyes, you can create “gates” in the data to exclude cell debris and potentially dead cells with low-to-no stain uptake.
Limitations
- Incomplete exclusion: Exclusion and viability dyes may not perfectly identify all dead cells, especially those in early stages of cell death.
- Background noise: Cellular debris and non-specific staining can mimic dead cell signatures, affecting accuracy.
- Varying sensitivity: The effectiveness of these techniques depends on the cell type, death mechanism, and assay design.
Can flow cytometry detect DNA?
Yes and no. It depends on what you mean by “detect DNA”. Flow cytometry can technically measure the amount of DNA present in a cell, but it cannot directly visualize or sequence the DNA itself.
Methods for measuring DNA content
- Fluorescent dyes: Specific dyes that bind to DNA, like propidium iodide (PI) or DAPI, are used. The intensity of the fluorescence signal is proportional to the amount of DNA present, allowing quantification.
- Cell cycle analysis: By measuring DNA content along with markers for other cell cycle phases, flow cytometry can determine the distribution of cells in different stages of cell division.
Limitations
- Cannot distinguish specific DNA sequences: These methods only give a bulk measurement of DNA content, not specific information about the DNA sequence or composition.
- Limited sensitivity: Detecting single copies of DNA, like in genomic DNA analysis, is challenging with standard flow cytometry due to background noise and limited signal amplification.
Alternative methods for DNA analysis
- Polymerase Chain Reaction (PCR): Amplifies specific DNA sequences for detection and analysis.
- Next-generation sequencing (NGS): Reads and sequences large amounts of DNA to identify sequences of interest.
- Fluorescence in situ hybridization (FISH): Visualizes specific DNA sequences in cells using fluorescent probes.
When is flow cytometry used for DNA analysis
- Measuring ploidy: Analyzing DNA content can reveal chromosomal abnormalities like aneuploidy (abnormal number of chromosomes).
- Cell cycle analysis: Understanding cell proliferation and cell cycle arrest is crucial in cancer research and other areas.
- Monitoring minimal residual disease (MRD): Detecting small numbers of cancer cells after treatment by measuring their DNA content.
Can flow cytometry detect apoptosis?
Yes, flow cytometry can be a valuable tool for detecting apoptosis, also known as programmed cell death. It offers several methods to assess different aspects of this process, each with its advantages and limitations.
Detecting DNA fragmentation
- Sub-G1 analysis: During apoptosis, the DNA gets fragmented into smaller pieces. By staining cells with dyes like propidium iodide (PI) and analyzing their DNA content using flow cytometry, a distinct peak appears to the left of the main G1 peak in the histogram. This “sub-G1” peak indicates the presence of apoptotic cells with fragmented DNA.
- TUNEL assay: This technique directly measures DNA fragmentation by labeling the exposed free ends of DNA with fluorescently tagged nucleotides. This method is highly specific for apoptosis but more complex to perform.
Assessing Annexin V binding
Annexin V is a protein that binds to phosphatidylserine, a phospholipid that gets flipped to the outer membrane surface during early apoptosis. Staining cells with Annexin V conjugated to a fluorescent dye allows identifying apoptotic cells in the early stages before DNA fragmentation occurs. Combining Annexin V with PI staining allows differentiating between early (Annexin V positive, PI negative) and late (both Annexin V and PI positive) apoptotic cells.
Measuring caspase activity
Caspases are enzymes that play a key role in executing apoptosis. Specific fluorescent probes can be used to measure caspase activity within individual cells by flow cytometry. This provides information about the activation and progression of the apoptotic pathway.
Mitochondrial membrane potential (MMP) analysis
During apoptosis, MMP decreases. Dyes like JC-1 can be used to assess MMP changes in individual cells, providing insights into the early stages of the apoptotic process.
Other considerations
- Flow cytometry cannot definitively diagnose apoptosis in isolation. Combining information from different methods and other cellular analyses is crucial for accurate interpretation.
- Sensitivity and specificity vary depending on the chosen technique and cell type. Optimization and appropriate controls are essential.
What are the advantages and disadvantages of flow cytometry?
Advantages of Flow Cytometry
- High-throughput analysis: Allows analyzing large numbers of cells (millions) quickly and efficiently compared to other methods like microscopy.
- Multiparameter analysis: Can measure multiple parameters simultaneously, including size, shape, surface markers, and even internal characteristics like DNA content, providing a comprehensive picture of individual cells.
- Sensitivity: Detects rare cell populations present in a complex sample, crucial for research and diagnosing diseases like leukemia.
- Single-cell analysis: Identifies and analyzes individual cells within a heterogeneous population, providing valuable insights into cellular diversity and function.
- Wide range of applications: Used in various fields like immunology, oncology, stem cell research, and drug discovery.
- Relatively non-invasive: Requires small sample sizes and minimal cell manipulation, making it suitable for studying delicate cell populations.
Disadvantages of Flow Cytometry
- High initial cost: Flow cytometers are expensive instruments, limiting accessibility for smaller labs or research groups.
- Technical expertise required: Operating and interpreting data requires specialized knowledge and training.
- Potential cell damage: Laser light and fluidics pressure can harm cells, demanding careful optimization and gentle handling practices.
- Limited information on intracellular details: While offering insights into internal characteristics like DNA content, it cannot directly visualize or analyze complex intracellular machinery or processes.
- Data complexity: Analyzing multiparameter data can be challenging and requires specialized software and expertise.
- Not a diagnostic tool on its own: Flow cytometry findings need integration with other clinical information and tests for accurate diagnosis.
Glossary of Related Medical Terms
- Antigen: A structural molecule, often a protein on the cell surface or inside the cell, recognized by specific antibodies.
- CD (Cluster of Differentiation): A standardized nomenclature system used to identify and investigate cell surface molecules (e.g., CD45 for leukocytes).
- Compensation: The mathematical process of correcting for the spectral overlap between different fluorochromes in multiparameter flow cytometry.
- Fluorochrome: A fluorescent chemical compound attached to an antibody that emits light at a specific wavelength when excited by a laser.
- Forward Scatter (FSC): Laser light scattered in the forward direction, which is proportional to the cell’s physical size.
- Gating: A computational method used in data analysis to select a specific sub-population of cells for further, isolated evaluation.
- Minimal Residual Disease (MRD): The minute number of malignant cells that remain in the patient during or after treatment, detectable by highly sensitive flow cytometry.
- Side Scatter (SSC): Laser light scattered at a 90-degree angle, which is proportional to the cell’s internal complexity or granularity.
Disclaimer: This protocol is intended for informational purposes only and may need to be modified depending on the specific laboratory procedures and patient circumstances. Always consult with a qualified healthcare professional for guidance. See additional information.
References
- Gorczyca W. Flow Cytometry in Neoplastic Hematology: Morphologic-Immunophenotypic-Genetic Correlation 4th Edition (2022). CRC Press.
- Goetz C, Hammerbeck C, Bonnevier J. Flow Cytometry Basics for the Non-Expert (Techniques in Life Science and Biomedicine for the Non-Expert) (2018). Springer.
- Ortolani C. Flow Cytometry Today: Everything You Need to Know about Flow Cytometry (2022). Springer.
- Porwit A, Bene MC. Multiparameter Flow Cytometry in the Diagnosis of Hematologic Malignancies (2018). Cambridge University Press.
- Leach M, Drummond M, Doig A, McKay P, Jackson B, Bain BJ. Practical Flow Cytometry in Haematology: 100 Worked Examples (2015). Wiley Blackwell.
- Ortolani C. Flow Cytometry of Hematological Malignancies 2nd Edition (2021). Wiley Blackwell.
- Kalodimou VE. Basic Principles in Flow Cytometry (2013). AABB Press.
- Hawley TS, Hawley RG. Flow Cytometry Protocols (Methods in Molecular Biology, 1678) (2018). Humana.